
Oly OA RNA isolation - juvenile whole body continued
Step 5: RNA Quality Check with Bioanalyzer (March 12, 2020)
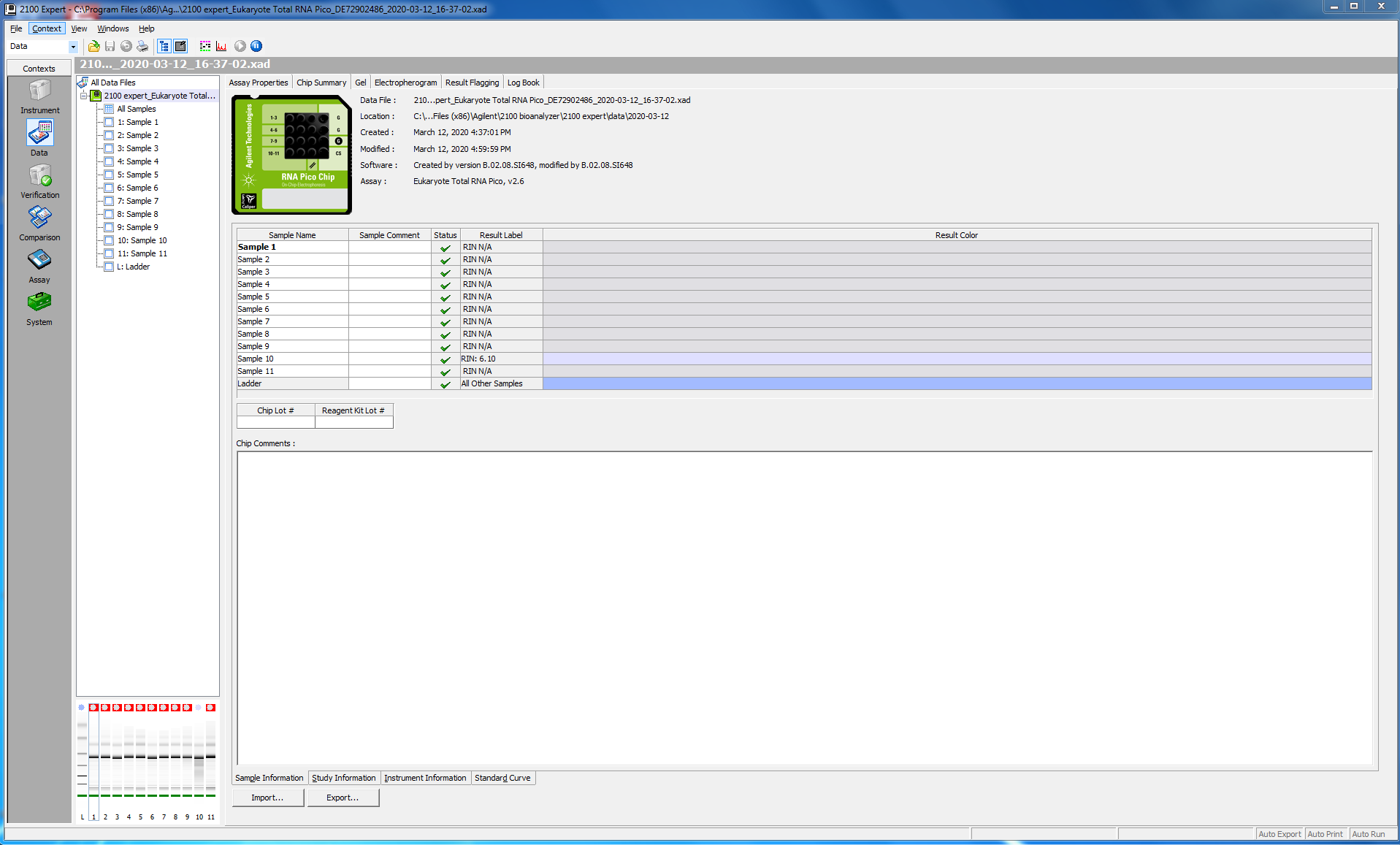
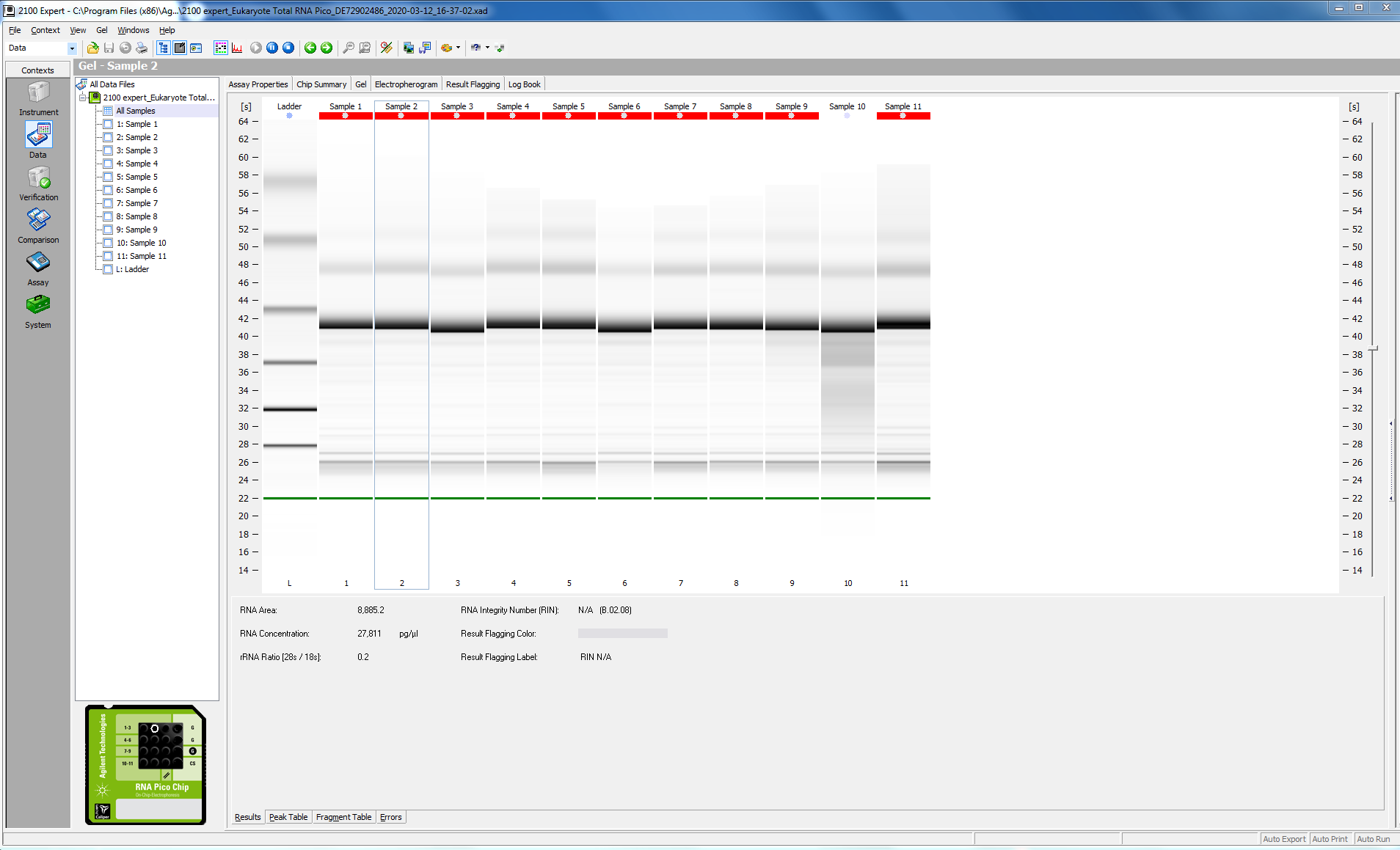
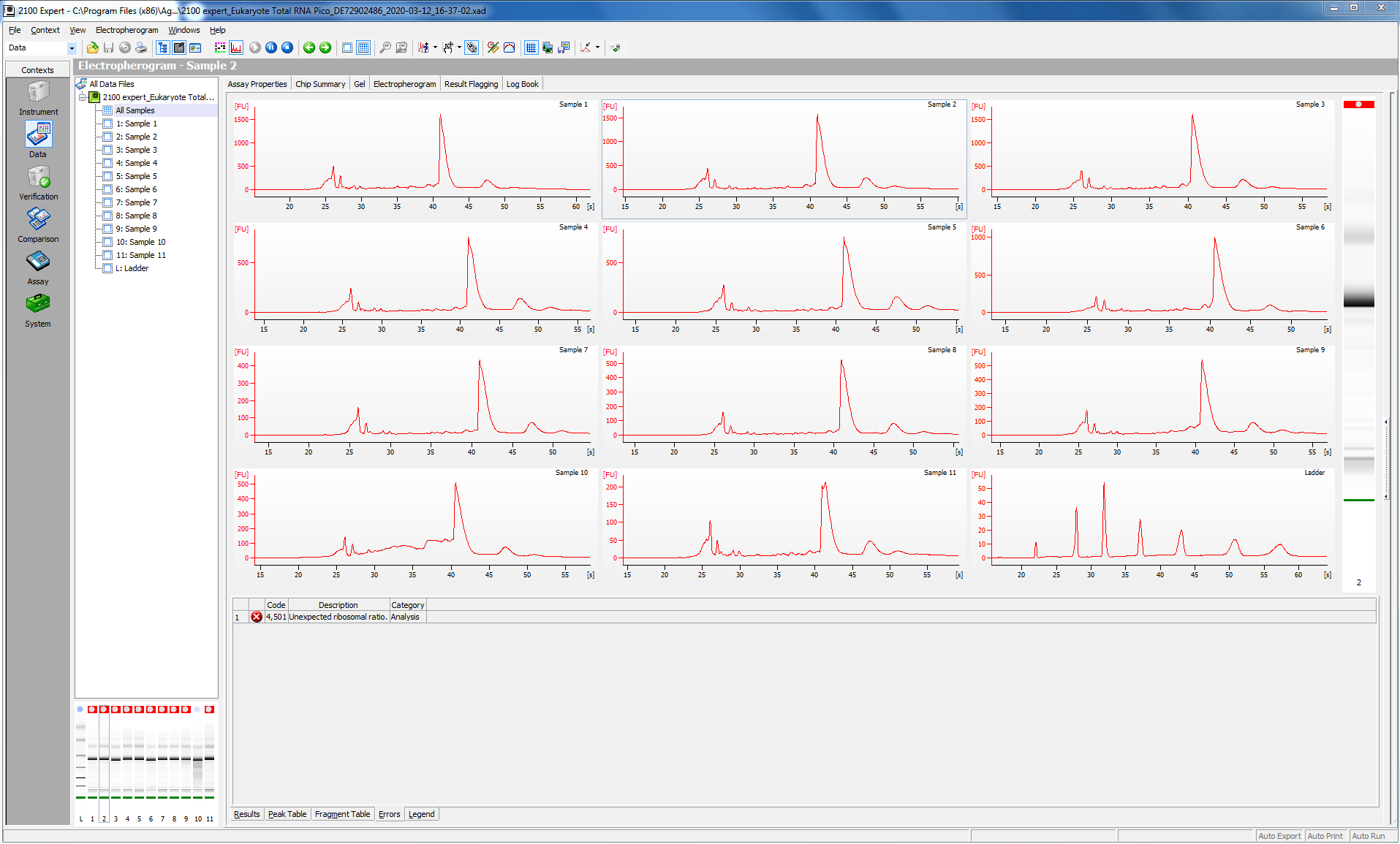
Before proceeding with the library prep, I checked the quality of my RNA (this was particularly critical, since I have just enough reagents to do my remaining 16 samples.
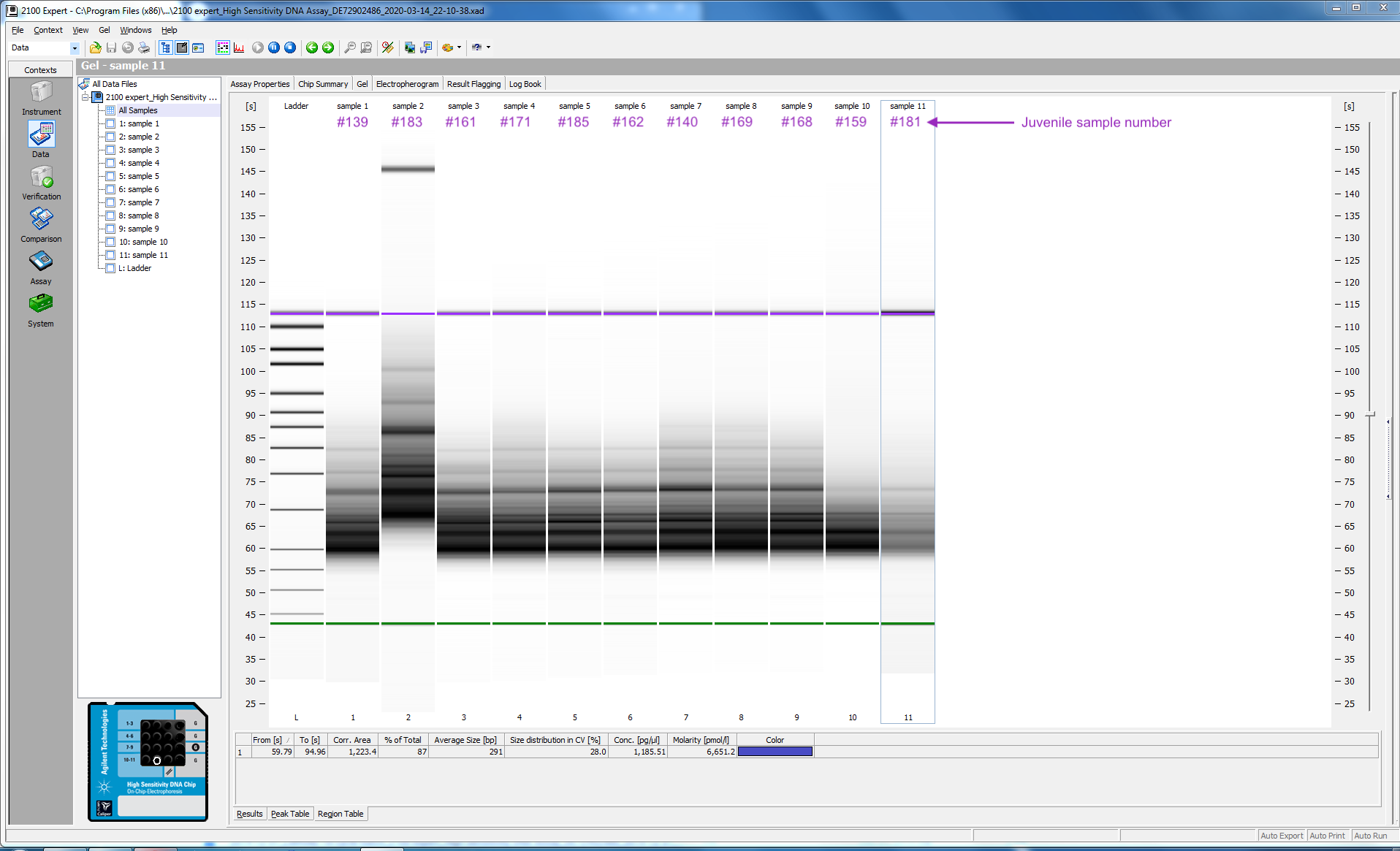
I tested 11 of the 16 samples; here is the bioanalyzer data file 2020-03-12_Juvenile-DNAsed-RNA_Pico-RNA.xad, and some screen shots from the run:
Step 6: Generate double stranded cDNA libraries (March 12, 2020)
I followed the QuantSeq protocol for standard input from first strand cDNA synthesis, and purification. Everything went smoothly.

Step 7: qPCR to assess optimal cycles (March 14, 2020)
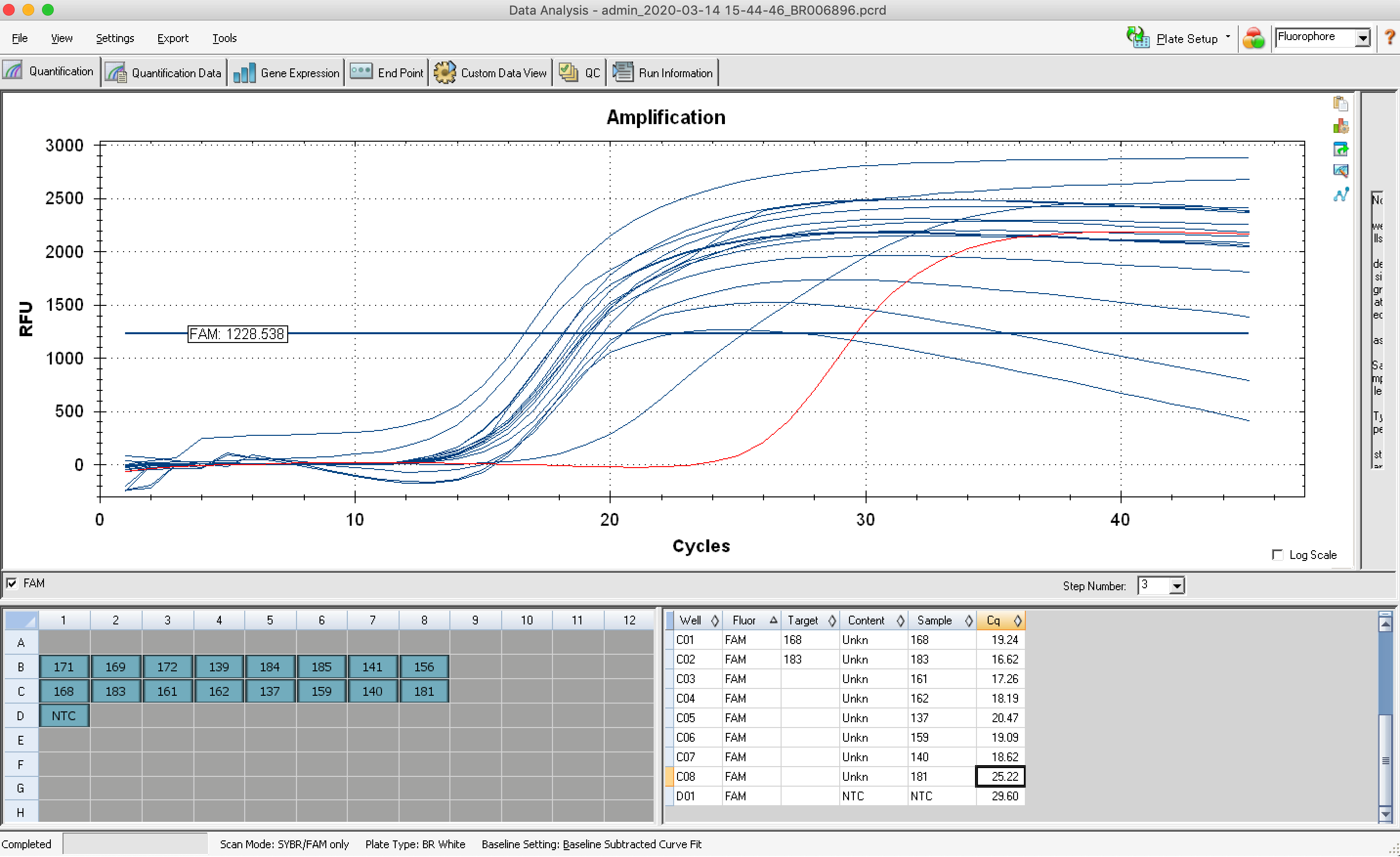
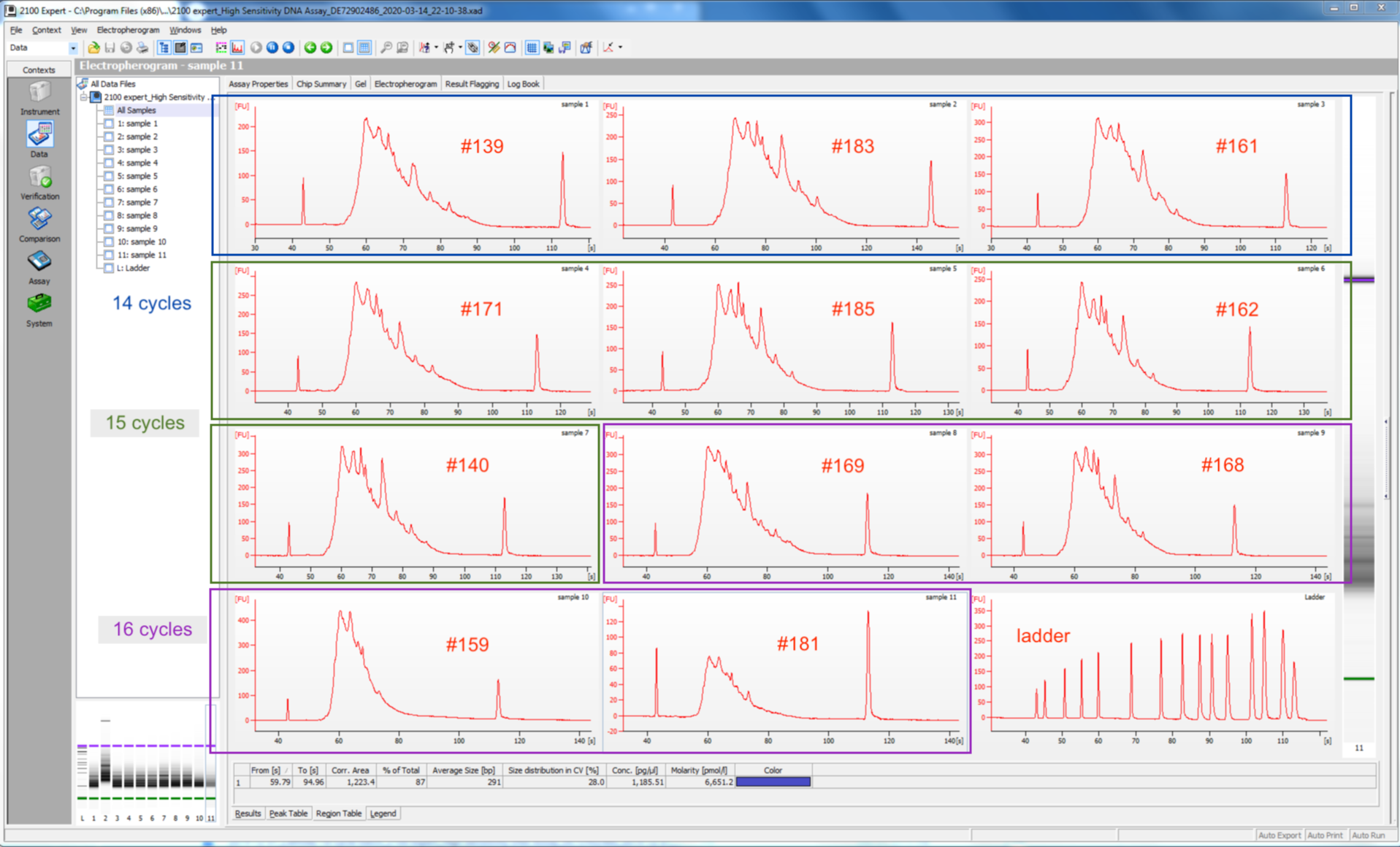
Before amplifying my libraries, I determined the optimal number of cycles for each sample via the qPCR add-on kit. I Ran into one issue - not enough primer solution. I had probably ~65% of what I needed. There was nothing I could do except move forward with the reduced primer concentration. I noticed that the amplification curves did not all plateau like normal - for some samples the RFU decreased after hitting their assumed peak RFU. I need to determine the optimal number of cycles based on the RFU at the plateau. For samples w/o plateaus, I used the maximum RFU value acheived. This resulted in all but one sample needing 14, 15, or 16 cycles. One sample (#181) needed 22 cycles (indicating very low amount of cDNA), but I instead ran it with the 16 cycle group, and figured I would check the resulting library quality and re-assess.
The resulting qPCR data file is located in this repo, Juvenile-whole-body_2020-03-14%2015-44-46_BR006896.pcrd, but also ALL qPCR data files are saved to the following folder on owl: https://owl.fish.washington.edu/scaphapoda/qPCR_data/cfx_connect_data/
qPCR protocol.

Amplification curves. In red is the NTC.
Step 8: Amplify libraries & purify (March 14, 2020)
I amplified my libraries in 3 batches, those needing 1) 14 cycles, 2) 15 cycles, and 3) 16 cycles (including the one sample neding 22 cycles). I performed the amplifications on the PTC 200 thermocycler. I then purified all the libraries at the same time. All went well.

Step 9: Assess cDNA library quality via Bioanalyzer (March 14, 2020)
I ran 11 of my completed libraries (from juvenile whole-body) on the Bioanalyzer using the Agilent High Sensitivity DNA chip and freshly prepared gel-matrix. Nearly all the libraries look great! Sample #181 looks like it has low concentration, and something weird may have happened with the #183. I’ll check with the lab to see if anyone has any ideas.
I might be done!
I’m debating whether to re-do sample #181, and possibly 183. If yes, then I have another day or two in the lab. If not, then I am done prepping my Quantseq libraries!
Getting quotes for sequencing
I’ve contacted several sequencing facilities for quotes. Here’s what I need and have learned in terms of sequencing setup:
- I have 144 libraries total. I indexed them in two discrete groups - within the groups they all have unique index numbers, but the two groups use the same numbers. So, they’ll need to at minimum be run on 2 lanes. Group 1 has 70 libraries, and Group 2 has 74 libraries.
- Do not run other libraries on the same lane as ours
- Some advice from Lexogen: “We recommend 3 – 5 M reads per sample for gene expression analysis. I recommend loading on the conservative side for PhiX and using 5%. Running Single Read chemistry is ideal here, 75, or 100bp is perfect.” (Although Katherine and her colleague both did 50bp, which seemed to work great).
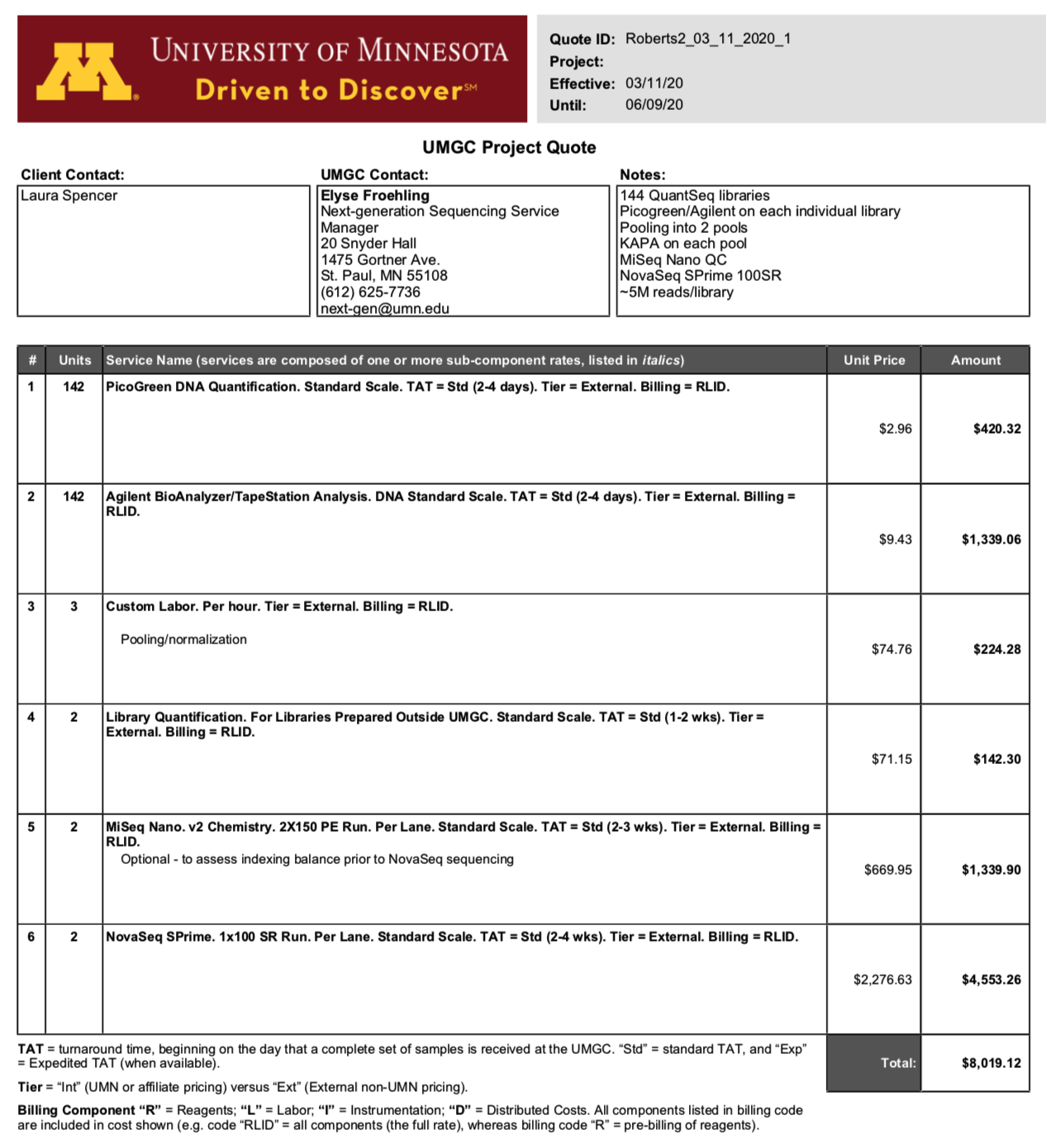
Univ. of Minnesota
“A quote is attached below. Note that for the last project we received the following information from Lexogen:
“We recommend 3 – 5 M reads per sample for gene expression analysis. I recommend loading on the conservative side for PhiX and using 5%. Running Single Read chemistry is ideal here, either 75 or 100bp is perfect.”
With this in mind, I’ve quoted you using 100SR sequencing at a depth of ~5M reads/sample. 100SR is a “custom” request for NovaSeq and is only available if you fill both lanes of the flow cell (which your project would). I included MiSeq Nano QC runs to assess barcode balance for each lane prior to loading on the NovaSeq. This is an optional but recommended step as it would allow us to make adjustments prior to loading on the NovaSeq.”
Univ. of Washington
“Since you will need <400M reads per lane to yield 3-5M reads per sample, I think NovaSeq SP will be the most cost effective option. The SP flow cell will generate ~800M single reads which we can split into two lanes each with ~400M reads. For reference, a HiSeq 4000 lane generates ~312M single reads.”
The 100 cycle flow cell I quoted is the smallest NovaSeq run that is available and could be run as either a single read 50bp or single read 75bp run. Another option would be a single read 75bp high output NextSeq run, but it generates 400k single reads (half the output of the NovaSeq run) and is only single input; this means we would not be able to split the flow cell into two lanes. In this case we would need to run two separate flow cells for your project. The 75 cycle NextSeq run is $1951 per flow cell as opposed to the 100 cycle NovaSeq run’s price of $3425. The 75 cycle NextSeq kit is the smallest version available.

Univ. of Texas, Austin
“Our default read-length is SR100; SR50 is hardly ever requested. Basically, you would have to use the entire flowcell for SR50 reads.”
Cost breakdown, but not accepting samples from external users at the moment
Univ. of California, Davis
Awaiting response
Novogene
For our HiSeq platform, we use PE150 for the strategy (we don’t offer other strategies.) The price would be $1,300 per lane and the data output is around 375M reads. As you suggested, two HiSeq lanes will be sufficient: giving 75 samples per lane for 5M reads. There is a $15 QC charge per sample (this would be charged one per pre-pooled library.) Finally, as a first time customer we can apply a $100 discount as well.