Isolated MORE RNA, Yaamini helps me with organization in R. Plan for tomorrow.
RNA Isolation
I did these samples:
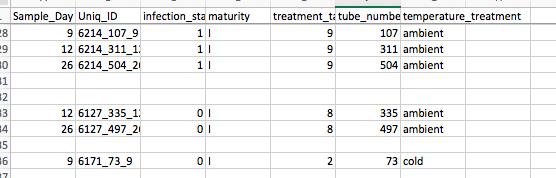
Tube 73, because I didn’t do it yesterday (accidentally did 37 instead)
Tubes 335 and 497 because they are the final two samples for the crab from tube 37 (FRP 6127)
Tubes 107, 311, and 504 because I needed a replacement infected, ambient crab
Qubit results:
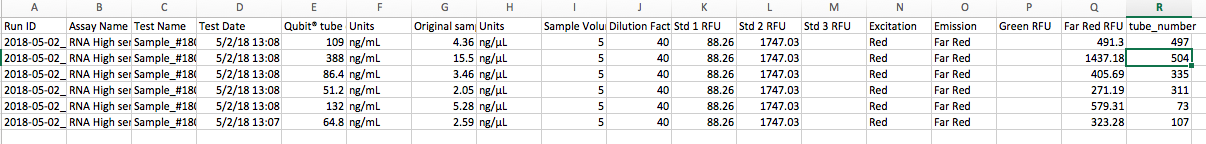
Tube 504 has a comparatively high RNA concetration (15.5 ng/µL). I will ask in the Crab Mtg #3 tomorrow if this is ok/ what it could mean.
Data organization using R
YAAMINI HELPED ME SO MUCH
So, what I wanted to do was to combine two files based on the common column, “tube_number”.
The files:
Qubit_consolidated.csv
- contains the Qubit results for ALL of the samples I have “isolated” RNA from
- contains all of the samples (from crabs that survived the experiment - three samples/crab) that I did the RNA isolation protocol on
I used this R script:
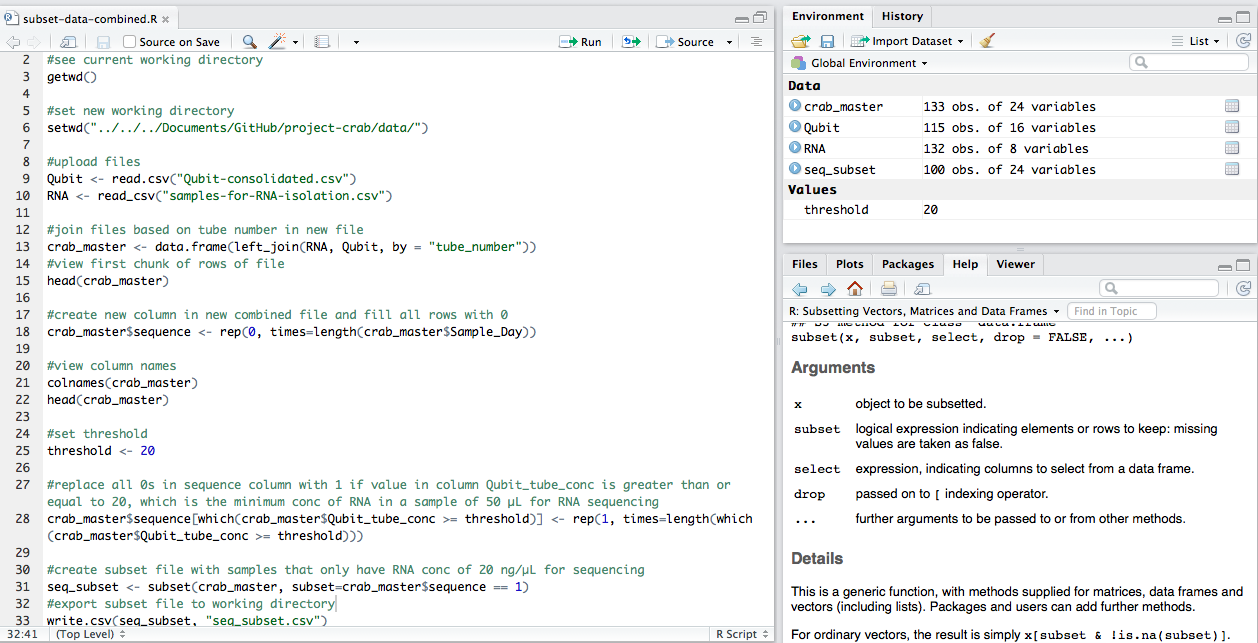
And I ended up with a new file that contains all of the samples that had a final RNA concentration (in 50µL of sample) of 20 ng/µL or higher:
Sample subset file
However,
The subset file has too many samples listed because it has some samples that are missing the third in the set (by set, I mean the three samples that each crab has). Also, it is excluding some of the samples from the warm treatment tanks. This is because some of the warm treatment tanks had “Out of range” or “0” RNA according to the Qubit.
I’m not sure if we want to send the warm treatment tank samples that have “Out of range” Qubit results. Will discuss during tomorrow morning’s meeting.
Tomorrow:
- Bioanalyzer with Sam on a couple samples (before meeting)
- 11am crab meeting
- work on data sheets, DecaPod FAQ Pt 2 and Crab Mtg #3