
Abstract
Altered cell metabolism enables tumours to sustain their increased energetic and biosynthetic needs. Although tumour metabolism has long been considered a promising discipline in the development of cancer therapeutics, the majority of work has focused on changes in glucose metabolism. However, the complexity of cellular metabolism means that very rarely is an individual metabolite required for a single purpose, and thus understanding the overall metabolic requirements of tumours is vital. Over the past 30 years, increasing evidence has shown that many tumours require glutamine as well as glucose for their proliferation and survival. In this minireview, we explore the complexity of glutamine metabolism in tumour cells, discussing how the overall context of the tumour dictates the requirement for glutamine and how this can affect the design of effective therapeutic strategies.
Similar content being viewed by others

Main
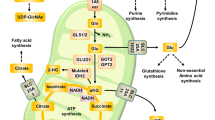
Altered metabolic pathways are commonly observed in almost all tumour types, resulting in different dependencies for specific nutrients or enzymes. Until recently, the majority of studies focused on changes in glucose flux, specifically the increased conversion of glucose to lactate (reviewed by Liberti and Locasale (2016)). The observation that mammalian cells rely on both glucose and glutamine (Reitzer et al, 1979; Moreadith and Lehninger, 1984; Board et al, 1990; Yuneva et al, 2007) shifted the focus to a more diverse range of pathways that are rewired in many tumours. Although glutamine is required as an alternative fuel for the tricarboxylic acid cycle (TCA) to produce ATP, it also contributes to a wide range of pathways in cells, producing amino acids, nucleotides and fatty acids as well as playing an important role in reactive oxygen species (ROS) homeostasis, mTOR activation and the hexosamine biosynthesis pathway (Figure 1). Current therapeutic approaches targeting glutamine metabolism focus on the initial stage of glutaminolysis by inhibiting glutaminase (Xiang et al, 2015). However, a greater understanding of the different roles that glutamine plays in tumours will aid the discovery of more specific therapeutic strategies against tumorigenesis.

Glutamine is required for multiple pathways in cells. Glutamine is converted to glutamate by glutaminase. Glutamate is then converted to αKG, which can be performed by GLUD to produce ammonia, which can regulate autophagy. Alternatively, this conversion can be performed through an aminotransferase reaction to produce an amino acid as well as αKG. This αKG can be used for both the forward and reverse fluxes of the TCA cycle, and can be used to regulate TET proteins, which alter DNA methylation. Bidirectional transport of glutamine and essential amino acids controls mTOR activation and autophagy regulation. Glutamine is used in the production of glutathione, which helps maintain the redox balance. Glutamine is also required for hexosamine biosynthesis and nucleotide biosynthesis.
In order to gain a greater understanding regarding the requirement for glutamine in different cancer settings, we need to consider how closely the models we use reflect the physiological situation in tumours. Much of the work done on glutamine metabolism in cancer has focused on the different pathways in vitro, whether in established cell lines or isolated tumour cells. The growth of cells in vitro varies considerably from the growth of cells in vivo, lacking the effects of the microenvironment and many of the cell–cell interactions that would occur in the tumour. Recent work has started to look at 3D cultures and growing cells in the presence of cancer-associated fibroblasts (CAFs) in order to recapitulate many of these features (Fiaschi et al, 2012; Asghar et al, 2015; Yang et al, 2016). However, more work needs to be done to study changes in tumour cell metabolism in vivo and where possible in human patients.
Glutamine addiction in cancer: pathways and products
Glutamine belongs to a unique class of amino acids that are thought of as ‘conditionally essential’. Glutamine can be synthesised as a scavenger for ammonia through the metabolism of other amino acids and so could be considered non-essential. However, under certain catabolically stressed conditions, such as sepsis, glutamine consumption increases markedly (Noguchi et al, 1997). Similarly, cells that are especially dependant on glutamine, such as those in the intestinal mucosa, rapidly undergo necrosis after glutamine deprivation (Lacey and Wilmore, 1990).
Likewise, specific cancer and oncogene-transformed cells are dependent on glutamine and undergo apoptosis during glutamine deprivation (Petronini et al, 1996; Yuneva et al, 2007; Weinberg et al, 2010). In these rapidly dividing cells, glutamine is avidly consumed and can act as a source for energy production, nitrogen and carbon for biomass, as well as being important in wider cell signalling.
Glutamine is an amido and amino group donor
Glutamine is first converted to glutamate by a glutaminase enzyme. It can then be converted to αKG in a number of ways, including using an isoform of glutamate dehydrogenase, either GLUD1 or GLUD2, to produce ammonia and αKG. In both human breast and lung cancer samples, increasing GLUD1 is associated with increasing malignant stage (Jin et al, 2015). Similarly, increased GLUD was associated with increased proliferation and invasion in colorectal carcinoma (Liu et al, 2015). However, glutamate can also produce αKG without producing ammonia through aminotransferase reactions.
Together with directing glutamine carbon into the TCA cycle (discussed below), aminotransferases are responsible for the use of glutamine’s amino group to produce amino acids, including alanine, aspartate and serine. The alanine aminotransferase enzymes (cytosolic GPT1 and mitochondrial GPT2) catalyse the reversible reaction of the amino group transfer from glutamate to pyruvate to produce αKG and alanine. This reaction plays an important role in the glucose–alanine cycle that is required to support gluconeogenesis in the liver. In cancer cells, GPT2 has been shown to couple increased glycolytic flux to increased glutamine catabolism in order to support the additional metabolic needs of the cells (Smith et al, 2016). Glutamine also supports the production of aspartate, using the cytosolic isoform of aspartate aminotransferase, GOT1. Aspartate is required for purine and pyrimidine synthesis as well as protein synthesis. Aspartate production was recently shown to be required for cell proliferation in the presence of electron transport chain (ETC) inhibition (Birsoy et al, 2015). The mitochondrial isoform, GOT2, operates with GOT1 as part of the Malate–Aspartate shuttle, which is required to shuttle electrons into the mitochondria for use in the ETC and to restore NAD+ pools required for glycolytic flux (Son et al, 2013).
Another aminotransferase that uses glutamate produced from glutamine is phosphoserine aminotransferase (PSAT), which is required for the de novo production of serine. Serine plays a vast number of different roles, from protein and nucleotide synthesis to acting as an allosteric activator to several different enzymes, such as PKM2 (Chaneton et al, 2012). It is also a precursor for a number of different metabolites, including glycine, sphingolipids and folate, which are required for the growth and survival of proliferating cells. In breast cancer cell lines, half of the αKG feeding into the TCA cycle was derived from PSAT activity, showing that serine biosynthesis can also supplement energy production in tumour cells (Possemato et al, 2011).
A recent study comparing quiescent and proliferating mammary epithelial cells demonstrated the important balance between various glutamine-consuming pathways within cells. Increased GLUD1 expression was associated with a quiescent state in these cells, whereas increased aminotransferase activity was seen in proliferating cells. Overexpression of GLUD1 simultaneously reduced cell proliferation and aminotransferase activity without altering aminotransferase expression levels (Coloff et al, 2016). Interestingly, although increased GLUD1 expression decreased mammary epithelial cell proliferation, the reverse occurred in colorectal carcinoma cells, where increased GLUD expression caused increased proliferation, migration and invasion (Liu et al, 2015). This highlights the changing importance of different metabolic pathways at different stages of both normal cell development and tumour progression as well as the variation in tumour metabolism between tissue types.
Glutamine does not just donate nitrogen to amino acids; it is also required as a nitrogen donor in the de novo synthesis of purines and pyrimidines, the nucleotide bases of DNA and RNA (reviewed by Lane and Fan (2015)). The amino nitrogen of glutamine is used to make nucleotide precursors, non-essential amino acids such as aspartate and glycine. The amido group is used to activate the ribose backbone, using PRPP amidotransferase, the first committed step of de novo purine synthesis, as well as to produce carbamoyl phosphate in the first step of pyrimidine metabolism.
Recent studies demonstrated that increased levels of glutamine synthetase (Stowell et al, 2015), an enzyme responsible for glutamine synthesis from glutamate and ammonia, promoted the enrichment of ammonia-derived nitrogen in de novo purine and pyrimidine synthesis. This supported the proliferation of human-transformed and cancer cell lines (Bott et al, 2015) as well as liver tumorigenesis in zebra fish (Cox et al, 2016). Interestingly, ectopic expression of the MYC oncogene has been shown to increase the expression of both glutaminase and GS promoting either glutamine catabolism or glutamine synthesis, respectively (Wise et al, 2008; Yuneva et al, 2012; Bott et al, 2015). Although upregulation of either of the activities can be context-specific and depend on cell requirements in a specific system, their co-existence in a same cell is also possible (Svenneby and Torgner, 1987). How the two processes are co-regulated and why the newly synthesised glutamine can be a preferred source for certain reactions with ample of exogenous glutamine available remain open questions.
Both carbon and nitrogen from glutamine are also used to produce proline. Proline is a non-essential amino acid required for protein biosynthesis, especially the production of the extracellular matrix protein collagen, which is known to provide protective mechanisms against chemotherapy in ovarian cancer (Choi et al, 2006). Likewise, the amido group of glutamine is used to convert aspartate to asparagine, which is essential in many tumour types (Krall et al, 2016; Yu et al, 2016).
Glutamine as a carbon source
One of the most important pathways glutamine supports is through the production of α-ketoglutarate (αKG), which enters into the TCA cycle. As well as being metabolised by oxidative decarboxylation, αKG produced from glutamate can also be metabolised by reductive carboxylation. Where the forward mode of the TCA cycle is required for energy production, under certain conditions, such as hypoxia (Le et al, 2012; Sun and Denko, 2014), or when mitochondrial respiration is impaired, the reductive carboxylation of glutamine-derived αKG into citrate is promoted (Fendt et al, 2013) and supports lipogenesis (Metallo et al, 2011; Mullen et al, 2011). This is especially important in tumours to sustain their increased requirement for cell membranes and cell signalling. These forward and reverse TCA cycle fluxes are not necessarily exclusive, which is frequently seen in cancer (Mcguirk et al, 2013), and although the direction of these fluxes are determined by the ratio of αKG to citrate (Fendt et al, 2013), the upstream determinants of this ratio are yet to be fully described.
ROS homeostasis
Glutamine is also involved in maintaining the redox balance. Reactive oxygen species can be both pro-tumorigenic, through ROS-mediated cell signalling, and highly damaging when in excess (reviewed by Liou and Storz (2010)). Reactive oxygen species are produced from several sources, including the mitochondrial ETC, which leaks electrons to oxygen to generate superoxide. Thus, increased glutamine oxidation can correlate with increased ROS production. However, a number of pathways involved in glutamine metabolism produce products that directly control ROS levels. For instance, glutamine is used to synthesise glutathione, a tripeptide (glu–cys–gly) that neutralises peroxide free radicals. Glutamine also regulates ROS homeostasis through the production of NADPH via GLUD, as well as producing NADPH using malic enzyme 1 (Son et al, 2013).
Glutamine regulates mTOR
The mTOR pathway, which can be regulated by glutamine, coordinates a wide variety of environmental cues to regulate cell growth and homeostasis, and is frequently deregulated in cancer. The availability of amino acids, including leucine, glutamine and arginine, stimulates mTOR activity independently of the activating mTOR mutations observed in cancer and so must be maintained regardless of mutation state (Sancak et al, 2008). The concentration of amino acids is partly regulated by obligatory amino-acid exchangers, including SLC38A1 (SNAT1), SLC38A2 (SNAT2), SLC1A5 (ASCT2) and SLC7A5 (LAT1), which co-ordinate the bidirectional transport of glutamine and leucine to regulate their concentrations within the cell (Broer et al, 2016), and thus partly regulate mTOR activity (Nicklin et al, 2009). However, these amino acids do not have to come from transporters, as macro-pinocytosis-derived amino acids can also support mTOR activation (Commisso et al, 2013).
Glutamine and autophagy regulation
The role of autophagy in cancer can be both tumour suppressive, by limiting oxidative stress and chromosomal instability, and pro-tumorigenic, by providing nutrients and supressing stress pathways. Many processes affected by glutamine metabolism suppress autophagy. For instance, glutamine concentrations regulate mTOR, which is known to inhibit autophagy, though as yet this mechanism is not fully understood (Dunlop and Tee, 2014). Similarly, ROS promote autophagy as a stress response, which can be prevented by glutamine, through the production of glutathione and NADPH. However, in some contexts, glutamine promotes autophagy. For instance, ammonia produced through the catabolism of glutamine by GLUD promotes autophagy (Cheong et al, 2011). The exact role of glutamine as an autophagy suppressor or activator appears to be context-dependent. For instance, some tumours convert glutamate to αKG via GLUD producing a second ammonium ion, whereas other tumours preferentially use an aminotransferase for this reaction, bypassing the production of this second ammonia, and thus suppressing autophagy.
Glutamine and glycosylation
Glutamine is also required for the hexosamine biosynthesis pathway, where it donates an amino group to glucose-6-phosphate to produce glucoseamine-6-phosphate. Through this pathway, both glucose and glutamine are required for O-linked and N-linked glycosylation, which is vital for both protein stability and function. Deregulated glycosylation is common in many tumour types (Stowell et al, 2015), occurring in both early and late cancer progression, and can be a result of changes in O- and N-glycan core structures or a result of changes in glycotransferase expression. Aberrant glycosylation has been observed to affect a number of oncogenes during tumorigenesis, including EGFR, where increased glycan branching results in increased residency time at the plasma membrane, thus increasing its active period (Lajoie et al, 2007). The co-ordination of glucose and glutamine metabolism, through the hexosamine biosynthesis pathway, has been shown to regulate signal transduction, through the glycosylation of the interleukin-3 receptor, which regulates cell growth and proliferation (Wellen et al, 2010). The ready availability of both nutrients is required for this pathway, ensuring sufficient energy production and biosynthesis, before activating signal transduction to promote cell growth.
Glutamine and epigenetic regulation
Epigenetic alterations are known to drive tumour progression. Many of these changes are regulated by pathways downstream of glutamine, for instance, DNA methylation can be reversed through the removal of oxidised methylated bases by TET proteins, which require αKG (Ito et al, 2011). A recent study found that areas of tumours with low glutamine concentrations have greater histone hypermethylation due to the decrease in αKG. Significantly, this resulted in BRAF inhibitor resistance in V600EBRAF melanoma cells (Pan et al, 2016), demonstrating the important link between tumour metabolism and epigenetic changes.
Glutamine addiction in cancer: thinking about therapeutics
Several glutamine metabolism-related therapeutic approaches are currently being developed. An inhibitor of GLS1, the kidney-specific isoform of glutaminase, CB-839, is currently in phase one clinical trials. As this drug targets the initial stage of glutamine catabolism, it should affect the functions downstream of glutamate. Compounds targeting glutamine transporters should also be effective against glutamine-dependent cancers, regardless of the specific need for glutamine, as they will target all pathways that require glutamine. However, targeting the precise step that the tumour requires could reduce wider toxicity and off-target effects, especially in healthy cells that also require glutamine for other pathways.
Isoform specificity
One way in which therapeutics against glutamine metabolism could be made more specific is by developing drugs against the particular isoforms of metabolic enzymes that are required in each tumour type, especially isoforms that are not commonly expressed in normal tissues. For instance, there are two isoforms of the glutaminase gene in mammals, kidney-type glutaminase (GLS1) and liver-type glutaminase (GLS2). GLS1 has two splice variants: KGA (the full-length variant) and GAC (which is missing the C-terminal). Expression of the more active isoform, GAC, is more frequently increased in several cancer types (Erickson and Cerione, 2010). Despite targeting both isoforms of GLS1, when CB-839 was tested against a panel of breast cancer cells, its effectiveness was limited to triple-negative breast cancer cells, which have elevated GAC expression and activity compared to receptor-positive cells (Gross et al, 2014).
Similarly, the expression and requirement of cytosolic and mitochondrial isoforms of aspartate and alanine aminotransferases are tissue- and tumour- specific. For instance, two recent studies have identified GPT2-mediated coupling of pyruvate production to glutamine catabolism in two different tissue types: PIK3CA-mutated colorectal cancer cells (Hao et al, 2016), and Ras- and p53-transformed YMAC cells (Smith et al, 2016). Although loss of GOT1, the cytosolic aspartate aminotransferase, kills leukaemia cells upon ETC inhibition (Birsoy et al, 2015), pancreatic ductal adenocarcinoma (PDAC) cells driven by mutant K-Ras rely on both GOT2, for aspartate production, and GOT1, to convert this aspartate into oxaloacetate. Oxaloacetate subsequently supports the reactions driven by cytosolic malate dehydrogenase and malic enzyme to produce NADPH and protect PDAC cells from oxidative stress (Son et al, 2013).
Combinatorial approaches
Some liposarcoma and breast cancer cells use asparagine as an exchange factor to import extracellular amino acids, and regulate mTOR activity and protein synthesis (Krall et al, 2016). Interestingly, these cells increase their dependence on exogenous asparagine once they have developed resistance to the GLS1 inhibitor, CB-839, suggesting that new therapeutic combinations against glutamine metabolism and its downstream effects may be effective.
Another example of a combinatorial approach targeting both glutamine catabolism and one of its downstream pathways was recently described. Here increased glutamine metabolism promoted resistance against mTOR inhibition in glioblastoma multiforme cell lines, with glutaminase expression increasing after mTOR kinase inhibition. However, combined mTOR and GLS1 inhibition resulted in massive synergistic tumour cell death and tumour inhibition (Tanaka et al, 2015). Combination therapies using multiple drugs: one targeting early-stage glutamine catabolism and one targeting the resulting downstream pathway could be an effective way to target glutamine-dependent tumours more specifically by limiting the emergence of compensatory pathways.
Oncogene involvement
Many oncogenes alter the expression of metabolic enzymes, including specific enzyme isoforms. For instance, K-Ras mutations can induce glutamine dependency, with different mutations determining the extent of glutamine dependence (Brunelli et al, 2014). Similarly, the c-MYC proto-oncogene is known to upregulate glutamine metabolism, specifically regulating GLS1, SLC1A5 and many of the genes involved in proline, nucleotide and serine biosynthesis (Wise et al, 2008; Liu et al, 2012). Conversely, in other systems, the c-MYC oncogene has also been shown to upregulate glutamine synthetase in order to promote glutamine production (Yuneva et al, 2012; Bott et al, 2015), demonstrating how c-MYC may activate glutaminolysis or glutamine synthesis in a context-dependent manner.
Tumour context and microenvironment
The requirement for glutamine in a range of different pathways means that understanding its increased catabolism in tumour cells is more complex than it first appears. Although glutamine is required for numerous tumour types, whether glutamine deprivation causes decreased cell proliferation or increased cell death varies between cell types (Rubin, 1990; Yuneva et al, 2007; Van Den Heuvel et al, 2012; Son et al, 2013). A greater understanding of the requirement for glutamine in specific systems is needed to identify novel therapeutic approaches and prognostic biomarkers.
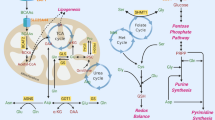
The role of glutamine in supporting the various pathways required for cell proliferation and survival has been extensively studied in in vitro systems, which do not take into account a complex tumour environment (Figure 2). A tumour’s metabolic requirements and their regulation can be changed upon cell adaptation to in vitro culture conditions, where the concentrations of glucose and glutamine are significantly higher than physiological, whereas other components are quite often missing. This notion was recently underscored by Davidson and co-authors, demonstrating that, although K-Ras-driven lung cancer cells in vitro are sensitive to glutaminase inhibition, K-Ras-driven lung tumours in vivo do not show the same dependency on glutamine and glutaminase (Davidson et al, 2016). Thus, this reflects how the tumour environment helps determine tumour metabolism, highlighting the importance of studying metabolic alterations in a physiological context.

2D in vitro cell culture lacks many of the features of the tumour microenvironment. The majority of in vitro cell culture systems involve growing a single cell type in a 2D monolayer, in nutrient-rich medium, and at fixed oxygen and carbon dioxide levels. In the tumour, cells grow in 3D, making cell:cell contacts and interact with a number of different cell types. The surrounding immune and stromal cells also affect the nutrient availability in the tumour microenvironment, as does proximity to blood vessels. Similarly, proximity to blood vessels also dictates the amount of oxygen that a cell receives.
Many tumours are genetically unstable, accumulating multiple mutations as they progress. These various mutations may arise in individual cells within the tumours and may promote different metabolic alterations. Thus, current studies that consider tumours as a whole may lose these subtleties in metabolic heterogeneity by averaging all changes across the whole tumour. Similarly, within tumours there are regions where nutrient and oxygen levels vary dependent on the proximity to blood vessels. This was recently demonstrated by Pan et al (2016) who show that low glutamine concentrations correlate with areas of low oxygen in xenograft tumours, resulting in histone hypermethlyation in these specific regions.
The effect of the microenvironment on tumours is well recognised. For instance, it is believed that epithelial cells can reprogram neighbouring stromal fibroblasts to increase the fibroblasts’ secretion of pyruvate and lactate, so that the epithelial cells may take up these energy-rich metabolites themselves (Pavlides et al, 2009). Similarly, CAFs undergo metabolic reprogramming to provide critical metabolites for their neighbouring tumour cells (Fiaschi et al, 2012). A recent study demonstrated that ovarian tumour cells reprogram their surrounding CAFs to produce more glutamine through glutamine synthetase than normal ovarian fibroblasts, enabling the cancer cells to fulfil their increased requirement for glutamine (Yang et al, 2016).
The tumour microenvironment is predominantly immunosuppressive. Immune cells in the tumour’s microenvironment are also known to adapt their metabolic phenotypes. For instance, the production of lactic acid by tumours, CAFs and macrophages results in an immunosuppressive phenotype in macrophages, aiding tumour progression (Ruan and Kazlauskas, 2013). Thus, it is vital that we understand the complex interplay between tumour cells and all the components of their environment, in order to truly understand the regulation of tumour metabolism and how best to combat tumour growth.
Concluding remarks
Metabolic alterations in tumour cells are more complex than first believed, where the increased uptake of a single amino acid could be required to support a wide range of different pathways, effecting energy production, cell signalling and ROS homeostasis. Similarly, the needs of one tumour vary greatly from another dependent on tissue type, oncogenic driver or micro-environmental support. Thus, the full context of tumour development and progression needs to be understood to develop therapies that will effectively treat tumours, with limited scope for resistance and off-target effects.
The complexity of glutamine metabolism means that the development of a ‘one-size fits all’ wonder drug against all glutamine-dependent tumours is unlikely. However, as we increase our understanding of the specific roles that glutamine plays in tumours, we edge ever closer to developing effective targeted therapies specific to each tumour.
References
Asghar W, El Assal R, Shafiee H, Pitteri S, Paulmurugan R, Demirci U (2015) Engineering cancer microenvironments for in vitro 3-D tumor models. Mater Today 18 (10): 539–553.
Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM (2015) An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162 (3): 540–551.
Board M, Humm S, Newsholme E (1990) Maximum activities of key enzymes of glycolysis, glutaminolysis, pentose phosphate pathway and tricarboxylic acid cycle in normal, neoplastic and suppressed cells. Biochem J 265 (2): 503–509.
Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D, Lu W, Pan J-A, Powers S, Rabinowitz J, Hatzoglou M, Murphy DJ, Jones R, Wu S, Girnun G, Zong W-X (2015) Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab 22 (6): 1068–1077.
Broer A, Rahimi F, Broer S (2016) Deletion of amino acid transporter ASCT2 (SLC1A5) reveals an essential role for transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to sustain glutaminolysis in cancer cells. J Biol Chem 291 (25): 13194–13205.
Brunelli L, Caiola E, Marabese M, Broggini M, Pastorelli R (2014) Capturing the metabolic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget 5 (13): 4722–4731.
Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH, Frezza C, O'reilly M, Gottlieb E (2012) Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491 (7424): 458–462.
Cheong H, Lindsten T, Wu J, Lu C, Thompson CB (2011) Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci USA 108 (27): 11121–11126.
Choi J, Credit K, Henderson K, Deverkadra R, He Z, Wiig H, Vanpelt H, Flessner MF (2006) Intraperitoneal immunotherapy for metastatic ovarian carcinoma: resistance of intratumoral collagen to antibody penetration. Clin Cancer Res 12 (6): 1906–1912.
Coloff JL, Murphy JP, Braun CR, Harris IS, Shelton LM, Kami K, Gygi SP, Selfors LM, Brugge JS (2016) Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab 23 (5): 867–880.
Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, Bar-Sagi D (2013) Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497 (7451): 633–637.
Cox AG, Hwang KL, Brown KK, Evason KJ, Beltz S, Tsomides A, O'connor K, Galli GG, Yimlamai D, Chhangawala S, Yuan M, Lien EC, Wucherpfennig J, Nissim S, Minami A, Cohen DE, Camargo FD, Asara JM, Houvras Y, Stainier DY, Goessling W (2016) Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat Cell Biol 18 (8): 886–896.
Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’brien JP, Pierce KA, Gui DY, Sullivan LB, Wasylenko TM, Subbaraj L, Chin CR, Stephanopolous G, Mott BT, Jacks T, Clish CB, Vander heiden MG (2016) Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab 23 (3): 517–528.
Dunlop EA, Tee AR (2014) mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 36: 121–129.
Erickson J, Cerione R (2010) Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget 1 (8): 734–740.
Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, Stephanopoulos G (2013) Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat Commun 4: 2236.
Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, Chiarugi P (2012) Reciprocal Metabolic Reprogramming through Lactate Shuttle Coordinately Influences Tumor-Stroma Interplay. Cancer Res 72 (19): 5130–5140.
Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, Mackinnon AL, Parlati F, Rodriguez ML, Shwonek PJ, Sjogren EB, Stanton TF, Wang T, Yang J, Zhao F, Bennett MK (2014) Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 13 (4): 890–901.
Hao Y, Samuels Y, Li Q, Krokowski D, Guan BJ, Wang C, Jin Z, Dong B, Cao B, Feng X, Xiang M, Xu C, Fink S, Meropol NJ, Xu Y, Conlon RA, Markowitz S, Kinzler KW, Velculescu VE, Brunengraber H, Willis JE, Laframboise T, Hatzoglou M, Zhang GF, Vogelstein B, Wang Z (2016) Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat Commun 7: 11971.
Ito S, Shen L, Dai Q, Wu S, Collins L, Swenberg J, He C, Zhang Y (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333 (6047): 1300–1303.
Jin L, Li D, Alesi G, Fan J, Kang H, Lu Z, Boggon T, Jin P, Yi H, Wright E, Duong D, Seyfried N, Egnatchik R, Deberardinis RJ, Magliocca K, He C, Arellano M, Khoury H, Shin D, Khuri F, Kang S (2015) Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 27 (2): 257–270.
Krall AS, Xu S, Graeber TG, Braas D, Christofk HR (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7: 11457.
Lacey J, Wilmore D (1990) Is glutamine a conditionally essential amino acid? Nutr Rev 48: 297–309.
Lajoie P, Partridge EA, Guay G, Goetz JG, Pawling J, Lagana A, Joshi B, Dennis JW, Nabi IR (2007) Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol 179 (2): 341–356.
Lane AN, Fan TW (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43 (4): 2466–2485.
Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJ, Lorkiewicz PK, Higashi RM, Fan TW, Dang CV (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15 (1): 110–121.
Liberti MV, Locasale JW (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41 (3): 211–218.
Liou G, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44 (5): 479–496.
Liu G, Zhu J, Cai C, Zhou Y, Yu M, Fu Z, Gong Y, Yang B, Li Y, Zhou Q, Lin Q, Ye H, Ye L, Zhao X, Li Z, Chen R, Han F, Tang C, Zeng B (2015) Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J Transl Med 13: 144.
Liu W, Le A, Hancock C, Lane A, Dang C, Fan T, Phang J (2012) Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcriiption factor c-MYC. Proc Natl Acad Sci USA 109 (23): 8983–8988.
Mcguirk S, Gravel S, Deblois G, Papadopol D, Faubert B, Wegner A, Hiller K, Avizonis D, Akavia U, Jones R, Giguere V, St-Pierre J (2013) PGC-1a supportsglutamine metabolism in breast cancer. Cancer Metab 1 (1): 22.
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G (2011) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481 (7381): 380–384.
Moreadith R, Lehninger A (1984) The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. J Biol Chem 259 (10): 6215–6221.
Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, Deberardinis RJ (2011) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481 (7381): 385–388.
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, Mackeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136 (3): 521–534.
Noguchi Y, James J, Fischer J, Hasselgren P (1997) Increased glutamine consumption in small intestine epithelial cells during sepsis in rats. Am J Surg 173 (3): 199–205.
Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, Hugo W, Song C, Xu X, Schones DE, Ann DK, Gradinaru V, Lo RS, Locasale JW, Kong M (2016) Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18 (10): 1090–1101.
Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP (2009) The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8 (23): 3984–4001.
Petronini PG, Urbani S, Alfieri R, Borghetti AF, Guidotti GG (1996) Cell susceptibility to apoptosis by glutamine deprivation and rescue: survival and apoptotic death in cultured lymphoma-leukemia cell lines. J Cell Physiol 169: 175–185.
Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun ZY, Cowley GS, Barretina J, Kalaany NY, Hsu PP, Ottina K, Chan AM, Yuan B, Garraway LA, Root DE, Mino-Kenudson M, Brachtel EF, Driggers EM, Sabatini DM (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476 (7360): 346–350.
Reitzer L, Wice B, Kennell D (1979) Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 254 (8): 2669–2676.
Ruan GX, Kazlauskas A (2013) Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem 288 (29): 21161–21172.
Rubin A (1990) Suppression of transformation by and growth adaptation to low concentrations of glutamine in NIH-3T3 cells. Cancer Res 50 (9): 2832–2839.
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320 (5882): 1496–1501.
Smith B, Schafer XL, Ambeskovic A, Spencer CM, Land H, Munger J (2016) Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep 17 (3): 821–836.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, Depinho RA, Cantley LC, Kimmelman AC (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496 (7443): 101–105.
Stowell SR, Ju T, Cummings RD (2015) Protein glycosylation in cancer. Annu Rev Pathol 10: 473–510.
Sun RC, Denko NC (2014) Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 19 (2): 285–292.
Svenneby G, Torgner IA (1987) Localization and function of glutamine synthetase and glutaminase. Biochem Soc Trans 15 (2): 213–215.
Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, Kyotani K, Mizowaki T, Imahori T, Ejima Y, Masui K, Gini B, Yang H, Hosoda K, Sasaki R, Mischel PS, Kohmura E (2015) Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest 125 (4): 1591–1602.
Van Den Heuvel AP, Jing J, Wooster RF, Bachman KE (2012) Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol Ther 13 (12): 1185–1194.
Weinberg F, Hamanaka R, Wheaton W, Weinberg S, Joseph J, Lopez M, Kalyanaraman, Mutlu G, Budinger G, Chandel N (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 107 (19): 8788–8793.
Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB (2010) The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24 (24): 2784–2799.
Wise DR, Deberardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, Mcmahon SB, Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105 (48): 18782–18787.
Xiang Y, Stine ZE, Xia J, Lu Y, O'connor RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG, Gao P, Sun L, Song L, Yan B, Slusher BS, Zhuo J, Ooi LL, Lee CG, Mancuso A, Mccallion AS, Le A, Milone MC, Rayport S, Felsher DW, Dang CV (2015) Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest 125 (6): 2293–2306.
Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, Han C, Baddour J, Marini JC, Ni J, Nakahara R, Wahlig S, Chiba L, Kim SH, Morse J, Pradeep S, Nagaraja AS, Haemmerle M, Kyunghee N, Derichsweiler M, Plackemeier T, Mercado-Uribe I, Lopez-Berestein G, Moss T, Ram PT, Liu J, Lu X, Mok SC, Sood AK, Nagrath D (2016) Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab 24 (5): 685–700.
Yu Q, Wang X, Wang L, Zheng J, Wang J, Wang B (2016) Knockdown of asparagine synthetase (ASNS) suppresses cell proliferation and inhibits tumor growth in gastric cancer cells. Scand J Gastroenterol 51 (10): 1220–1226.
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y (2007) Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 178 (1): 93–105.
Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, Chen X, Bishop JM (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15 (2): 157–170.
Acknowledgements
The Yuneva lab is supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001223), the UK Medical Research Council (FC001223) and the Wellcome Trust (FC001223).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Still, E., Yuneva, M. Hopefully devoted to Q: targeting glutamine addiction in cancer. Br J Cancer 116, 1375–1381 (2017). https://doi.org/10.1038/bjc.2017.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.113
Keywords
This article is cited by
-
Glutamine metabolism-related genes and immunotherapy in nonspecific orbital inflammation were validated using bioinformatics and machine learning
BMC Genomics (2024)
-
ARPC1A correlates with poor prognosis in prostate cancer and is up-regulated by glutamine metabolism to promote tumor cell migration, invasion and cytoskeletal changes
Cell & Bioscience (2023)
-
Bioinformatics and machine learning were used to validate glutamine metabolism-related genes and immunotherapy in osteoporosis patients
Journal of Orthopaedic Surgery and Research (2023)
-
Small molecule inhibitors for cancer metabolism: promising prospects to be explored
Journal of Cancer Research and Clinical Oncology (2023)
-
Functional enrichment analysis of LYSET and identification of related hub gene signatures as novel biomarkers to predict prognosis and immune infiltration status of clear cell renal cell carcinoma
Journal of Cancer Research and Clinical Oncology (2023)
