Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain


1
Department of Biofunctional Analysis Laboratory of Molecular Biology, Gifu Pharmaceutical University, 1-25-4 daigaku-nishi, Gifu 501-1196, Japan
2
Department of Pharmacology, Graduate School of Pharmaceutical Sciences, Tohoku University, 6-3 Aramaki-Aoba, Aoba-ku, Sendai, Miyagi 980-8578, Japan
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(1), 20; https://doi.org/10.3390/ijms19010020
Submission received: 23 November 2017
/
Revised: 19 December 2017
/
Accepted: 20 December 2017
/
Published: 22 December 2017
(This article belongs to the Special Issue Kinase Signal Transduction 2017)
{kind=link}
Abstract
:Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII), a multifunctional serine (Ser)/threonine (Thr) protein kinase, regulates diverse activities related to Ca2+-mediated neuronal plasticity in the brain, including synaptic activity and gene expression. Among its regulators, protein phosphatase-1 (PP1), a Ser/Thr phosphatase, appears to be critical in controlling CaMKII-dependent neuronal signaling. In postsynaptic densities (PSDs), CaMKII is required for hippocampal long-term potentiation (LTP), a cellular process correlated with learning and memory. In response to Ca2+ elevation during hippocampal LTP induction, CaMKIIα, an isoform that translocates from the cytosol to PSDs, is activated through autophosphorylation at Thr286, generating autonomous kinase activity and a prolonged Ca2+/CaM-bound state. Moreover, PP1 inhibition enhances Thr286 autophosphorylation of CaMKIIα during LTP induction. By contrast, CaMKII nuclear import is regulated by Ser332 phosphorylation state. CaMKIIδ3, a nuclear isoform, is dephosphorylated at Ser332 by PP1, promoting its nuclear translocation, where it regulates transcription. In this review, we summarize physio-pathological roles of CaMKII/PP1 signaling in neurons. CaMKII and PP1 crosstalk and regulation of gene expression is important for neuronal plasticity as well as survival and/or differentiation.
1. Introduction
Protein phosphorylation, one of the most important post-translational modifications, drives rapid, reversible and extracellular signal-dependent cell signaling. In the brain, Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII), a multifunctional serine (Ser) and threonine (Thr) kinase [1], regulates diverse Ca2+-mediated neuronal activities, including neurotransmitter release, gene expression, and synaptic plasticity [2,3]. CaMKII is a dodecameric holoenzyme assembled from α, β, γ, and δ isoforms. In eukaryotes, these four CaMKII isoforms are encoded by distinct genes, and their corresponding mRNAs are alternatively spliced to give rise to subtypes exhibiting variable domains [4,5].
CaMKII has attracted substantial attention due to its function in synaptic plasticity, an activity that occurs at postsynaptic densities (PSDs) [6,7,8]. In response to Ca2+ elevation by extracellular stimuli, Ca2+-CaM binding to CaMKII displaces autoinhibitory domains to allow ATP and exogenous substrates access to the active site. Immediately after activation, Thr286 in the autoinhibitory domain of the α isoform (corresponding to Thr287 of β, γ, and δ isoforms) is autophosphorylated by the neighboring kinase domain. This event increases Ca2+-CaM binding affinity and blocks interaction of autoinhibitory and catalytic domains, thereby generating autonomous kinase activity and prolonging the Ca2+/CaM-bound state. CaMKIIα autonomy is critical for induction and maintenance of hippocampal long-term potentiation (LTP), both of which underlie learning and memory [9,10].
The 12 subunits of the CaMKII holoenzyme assemble into two coplanar rings, each containing six subunits [11]. These ring structures suggest a potential mechanism for establishment of an autonomous kinase state, as it is proposed that autophosphorylation occurs by an inter-subunit process [12]. Indeed, following a robust and long Ca2+ stimulus, two adjacent CaMKII monomers are simultaneously bound by Ca2+/CaM. In these conditions, one subunit serves as a substrate for the other, resulting in Thr286/Thr287 phosphorylation. Once the first subunit is phosphorylated, subsequent phosphorylation within the holoenzyme is more likely to occur, as lower Ca2+ levels are required for the second phosphorylation. Thus, in this scenario, CaMKII remains active, even when Ca2+ levels return to basal levels, until it is dephosphorylated. If the number of phosphorylated subunits exceeds a threshold and the phosphorylation rate is greater than the dephosphorylation rate, then CaMKII activity is sustained [13].
In contrast to postsynaptic CaMKII function, the physiological relevance of nuclear activity of CaMKII isoforms in the central nervous system (CNS) remains unclear. Alternative splicing of CaMKII generates a multitude of isoforms for each CaMKII subunit [5]. Among these alternatively splicing isoforms, CaMKIIαB [14], CaMKIIγA [15], and CaMKIIδ3 (also called CaMKIIδB) [16] display consensus (KKRK) sequences in respective variable domains that resemble a nuclear localization signal (NLS) and are homologous to the simian virus 40 (SV40) large T antigen NLS [17]. In rat brain neurons, CaMKIIαB and CaMKIIδ3 are expressed in the nucleus [14,18], and their activity is reportedly regulated by the NLS motif, which, when phosphorylated, prevents nuclear localization. CaMKIIδ3 Ser332, which is immediately C-terminal to the NLS (328KKRKS332), is reportedly phosphorylated by the CaMK family members CaMKI or CaMKIV, blocking association of CaMKII with the NLS receptor m-pendulin and thereby preventing nuclear localization [19].
The Ser/Thr phosphatase Protein Phosphatase-1 (PP1), a key regulator of CaMKII signaling, forms a heterodimer comprised of a catalytic (PP1c) and a regulatory subunit. PP1c can form a complex with over 50 regulatory or scaffolding proteins that dictate substrate specificity and subcellular localization [20]. In mammalian cells, PP1c itself occurs as different isoforms (α, β, δ, γ1 and γ2) [21,22,23,24,25], and three (PP1α, PP1β, and PP1γ1) are highly expressed in the brain [26]. All isoforms show nearly 90% amino acid homology and are most divergent at the N- and C-termini. Importantly, although CaMKIIα Thr286 can be dephosphorylated by PP1, PP1 appears to play a more prominent role in CaMKII dephosphorylation at PSDs [27]. Moreover, CaMKIIδ3 is dephosphorylated at Ser332 by PP1, promoting its nuclear translocation [28].
In this review, we focus on the role of CaMKII/PP1 signaling in both neuronal plasticity at PSDs and gene expression in the nuclei. We also discuss how imbalanced CaMKII/PP1 activity may underlie neuronal pathologies, such as mental disorders and neurodegeneration.
2. Physiological Function of CaMKII/PP1 Signaling at PSDs
PSDs are localized in the tips of dendritic spine heads and contain multiple classes of proteins that function in neuronal signaling in response to presynaptic neurotransmitter release, such as glutamate [29]. CaMKII is one of the most abundant proteins found in forebrain PSDs [30]. CaMKII regulates synaptic strength, in part by phosphorylating glutamate receptors [31]. As noted, CaMKIIα Thr286 autophosphorylation promotes autonomous kinase activity, which when sustained is essential for learning and memory [9,10]. Thus, it is critical to understand how CaMKII remains highly phosphorylated and resists endogenous phosphatase activity.
One factor governing this persistent “on-state” is PP1 localization to dendritic spines and PSDs [32,33]. PP1 inhibition enhances CaMKIIα Thr286 autophosphorylation during LTP induction [34]. However, autophosphorylated CaMKIIα Thr286 cannot be dephosphorylated by PP1 in purified PSDs from rats [35]. These authors also showed that the Thr286 site is not buried within the CaMKIIα protein, as it can be dephosphorylated in purified PSDs by exogenous soluble PP1c or λ phosphatase. These results indicate that the inability of PP1 to dephosphorylate this site in vivo is due to the positioning of PP1 in PSDs and the inhibitory activity of scaffolding proteins that modulate PP1 activity [36,37]. For example, spinophilin and its homolog neurabin are F-actin binding proteins that target PP1 to PSDs, and spinophilin alters PP1 catalytic activity by steric inhibition of substrate binding sites [38]. Indeed, numerous protein-protein interactions hold PP1 in such a position that PP1 simply cannot reach CaMKIIα Thr286.
Once activated, CaMKII remains in an active conformation throughout the LTP maintenance phase, an observation that forms the basis of the hypothesis that CaMKII is critical for memory formation [9,10]. However, these conclusions are based on work carried out using hippocampal homogenates, and these studies do not provide specific information relevant to the pool of active CaMKII at synapses. Some studies using Camui, a fluorescence resonance energy transfer (FRET)-based CaMKII sensor [39], show that CaMKII activity lasts only ~1 min after stimulation during LTP induction, based on two-photon laser-mediated photolysis of caged glutamate at hippocampal CA1 spines [40,41]. CaMKII activity, as measured by the magnitude of the Camui-FRET change, was not affected by treatment with Calyculin A, a PP1/PP2A phosphatase inhibitor [41]. Thus, optical monitoring of CaMKII activity has the advantage of greater spatiotemporal resolution over previous immunoblotting studies. However, they still have technical limitations relating to their ability to detect small amounts of activated CaMKII within dendritic spines. Thus, the relationship between LTP and CaMKII/PP1 signaling needs further investigation.
3. Pathological CaMKII/PP1 Signaling in PSDs
In animal models of Parkinson’s disease, striatal dopamine depletion increases CaMKIIα autophosphorylation at Thr286 in parallel with decreased PP1γ1 activity and increased PP1γ1 binding to spinophilin [42,43,44]. Moreover, we showed increased CaMKIIα Thr286 autophosphorylation and decreased levels of spinophilin and PP1 in the prefrontal cortex of a mouse model of α-thalassemia X-linked mental retardation (ATR-X) syndrome [45]. This pathological imbalance of CaMKII/PP1 signaling in the ATR-X model correlated with altered dendritic spine morphology, suggesting that CaMKII/PP1 signaling regulates this process [45]. Likewise, decreased PP1 activity in the brain of Angelman syndrome model mice correlated with increased phosphorylation of hippocampal CaMKIIα at Thr286 in PSDs, as well as with changes in synaptic plasticity, learning, and memory [46]. This evidence suggests overall that increased CaMKII activity is mediated by reduced PP1 activity, particularly in PSDs, thereby perturbing synaptic plasticity and learning and memory.
4. Physiological CaMKII/PP1 Signaling in Nuclei
Transduction of signals from synapses to the nucleus is primarily mediated by Ca2+ signaling, and nuclear Ca2+ transients are some of the most potent regulators of neuronal gene expression [47]. Nuclear CaMKII transcriptionally regulates the gene encoding neurotrophin brain-derived neurotrophic factor (BDNF) [48,49] through phosphorylation of diverse nuclear proteins, including cAMP response element-binding protein (CREB) [50,51], methyl CpG binding protein 2 (MeCP2) [52], activating transcription factor [53,54], CCAAT/enhancer-binding protein [55,56], and serum response factor [57].
Specifically, CaMKII phosphorylates CREB at Ser133 and Ser142 in vitro [51]. Moreover, Ca2+-induced CaMKII activation in primary cultured neurons stimulates CREB phosphorylation at Sers 133, 142, and 143 [58]. CREB phosphorylation at Ser142 and Ser143 contributes to its activation, and alanine mutations at Ser142 and Ser143 block Ca2+-induced CREB-dependent transcription [58]. However, transgenic mice harboring a single CREB Ser142-to-alanine mutation show alterations in the circadian clock located in the suprachiasmatic nucleus, which down-regulate c-Fos, a transcriptional target of CREB [59]. The transcription factor MeCP2 binds to methylated cytosine residues of CpG dinucleotides in DNA [60]. Neuronal activity and subsequent Ca2+ influx trigger CaMKII-dependent MeCP2 phosphorylation at Ser421 [52]. Knock-in mice that lack MeCP2 Ser421 or Ser421 and Ser424, a second site of synaptic activity-induced phosphorylation, show perturbed synaptogenesis, synaptic plasticity, and spatial memory [61,62], underscoring the importance of these phosphorylation sites in vivo.
Until recently, mechanisms underlying substrate phosphorylation by nuclear CaMKII remained unclear. Thus, we investigated nuclear-cytoplasmic shuttling of the nuclear isoform CaMKIIδ3. Previously, others had reported that CaMKIIδ3 Ser332, which is C-terminal to the NLS (328KKRKS332), is phosphorylated by CaMKI or CaMKIV, prohibiting nuclear localization [19]. To investigate a potential function of CaMKII phosphorylation, we generated a specific antibody against phosphorylated Ser332 of CaMKII. In an in vitro phosphorylation assay of purified rat brain CaMKII, CaMKIIδ3 was dephosphorylated by PP1 at both Ser332 and Thr287 [28]. We also showed that PP1α and PP1γ1 predominantly regulate CaMKIIδ3 nuclear translocation in Neuro-2a cells. However, nuclear CaMKIIδ3 activity in Neuro-2a cells was enhanced by PP1γ1 overexpression. Consistent with these results, in experiments using primary cultured mesencephalic dopamine neurons, CaMKIIδ3 was dephosphorylated only at Ser332, not at Thr287, by activated PP1 [28]. This discrepancy may be explained by the binding of various proteins to the CaMKII/PP1 complex, in a manner similar to spinophilin in PSDs. We conclude that the in vitro experimental conditions used in our study resemble the cytosolic microenvironment, in which PP1 directly dephosphorylates cytosolic CaMKIIδ3. We have not yet defined proteins binding to and regulating the CaMKIIδ3/PP1 complex in vivo, an analysis that awaits future studies.
Others have reported nuclear activity of CaMKIIαB and CaMKIIγA in neurons [63,64]. For example, in rat retinal ganglion cells CaMKIIαB expression and nuclear translocation increase via an unknown mechanism following glutamate-induced cell death [63]. Ma et al. also reported that CaMKIIγA functions as a transporter of Ca2+/CaM to the nucleus following depolarization of cultured superior cervical ganglion neurons and that the Ca2+/CaM-CaMKIIγ complex is dephosphorylated at Ser334 by calcineurin, allowing it to shuttle to the nucleus. Nuclear delivery of Ca2+/CaM activates nuclear CaM kinases, including CaMKIV and CaMKK, driving CREB phosphorylation and transcription of its target genes [64]. Therefore, phosphatases other than PP1, such as calcineurin and/or PP2A, may dephosphorylate Ser332 of CaMKIIδ3 in other types of neurons.
5. Pathological CaMKII/PP1 Signaling in Nuclei
CaMKII-PP1 signaling transcriptionally regulates BDNF, a factor vital for neuronal survival, growth, and maintenance, in brain circuits functioning in emotion and cognition [65]. MeCP2 mutations cause most cases of Rett syndrome, an X-linked dominant neurodevelopmental disorder and a leading cause of mental retardation and autistic behavior in females [66]. Phenotypes, such as normal early development followed by progressive motor and cognitive dysfunction, seen in mice that either lack or overexpress MeCP2 recapitulate many characteristic features of Rett syndrome [67,68,69]. In addition, like syndrome patients, MeCP2 mutant mice show abnormalities in brain morphology and cyto-architecture, in particular a decrease in dendritic arborization and spine loss [52,70]. Importantly, MeCP2 Ser421 phosphorylation by CaMKII is required for activity-dependent regulation of BDNF gene expression [52], suggesting that transcriptional deregulation of this gene potentially due to CaMKII dysregulation plays a central role in Rett syndrome.
We also previously revealed that nuclear CaMKII/PP1 signaling is important for neuronal survival and differentiation [28]. We reported that the nuclear isoform CaMKIIδ3 is highly expressed in dopaminergic rat substantia nigra neurons [71] and that stimulation of the dopamine D2 receptor (D2R) activates CaMKIIδ3, inducing BDNF gene expression in NG108-15 cells [72]. We also found that CaMKIIδ3 Ser332 is directly dephosphorylated by PP1, promoting CaMKIIδ3 nuclear translocation, and that aripiprazole (APZ), a dopamine D2R partial agonist, promotes CaMKIIδ3 nuclear translocation and enhances BDNF expression [28]. APZ treatment also enhanced sprouting and survival of cultured dopaminergic neurons through the CaMKIIδ3/PP1 pathway [28]. Consistent with our results, APZ treatment for eight weeks was reported to significantly increase plasma BDNF levels in first-episode untreated schizophrenia patients [73]. BDNF protein expression decreases in the dopamine-deficient substantia nigra of Parkinson disease patients [74,75]. BDNF also reportedly promotes survival of cultured mesencephalic dopaminergic neurons [76] and, in vivo, protects dopaminergic neurons from damage by the neurotoxins 1-methyl-1,2,3,6-tetrahydropiridine and 6-hydroxydopamine [77]. This evidence and our data suggest a critical role for BDNF in supporting survival and/or differentiation of midbrain dopaminergic neurons functioning nuclear CaMKII/PP1 pathway with the APZ treatment.
6. Conclusions
CaMKII/PP1 signaling plays a crucial role in many different aspects of synaptic plasticity in PSDs and in activity-regulated transcription in nuclei. CaMKII alternative splicing generates numerous subtypes of each CaMKII isoform. Figure 1 summarizes how each function, in relationship to others, mediates Ca2+ signaling to PSDs or nuclei. However, the composition of the dodecameric CaMKII holoenzyme affects CaMKII localization [78,79]. The ability of CaMKII to translocate to the nucleus is thus governed by the presence of nuclear versus cytoplasmic isoforms that make up holoenzyme [17]. Nuclear CaMKII isoforms containing an NLS (CaMKIIαB, CaMKIIδ3, and CaMKIIγA) may co-assemble with cytoplasmic subunits, including postsynaptic density-associated CaMKIIα [80] and/or F-actin-associated CaMKIIβ [81] to facilitate synaptic activation or nuclear translocation. Further study is required to reveal the relationship between oligomerization of heterogenous CaMKII isoforms and PP1 in neurons.
Acknowledgments
This work was supported by MEXT/JSPS KAKENHI Grant 25460090 (to Norifumi Shioda).
Author Contributions
Norifumi Shioda and Kohji Fukunaga wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AC | adenylate cyclase |
| ATR-X | α-thalassemia X-linked mental retardation |
| BDNF | brain-derived neurotrophic factor |
| CaM | calmodulin |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| cAMP | cyclic adenosine monophosphate |
| CNS | central nervous system |
| CREB | cAMP response element-binding protein |
| D2R | dopamine D2 receptor |
| LTP | long-term potentiation |
| MeCP2 | methyl CpG binding protein 2 |
| NLS | nuclear localization signal |
| NMDA | N-methyl-d-aspartate |
| PKA | protein kinase A |
| PP1 | Protein Phosphatase-1 |
| PSDs | postsynaptic densities |
| Ser | serine |
| SV40 | simian virus 40 |
| Thr | threonine |
| VDCC | voltage-dependent calcium channel |
References
- Braun, A.P.; Schulman, H. The multifunctional calcium/calmodulin-dependent protein kinase: From form to function. Annu. Rev. Physiol. 1995, 57, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Colbran, R.J.; Soderling, T.R. Calcium/calmodulin-dependent protein kinase II. Curr. Top. Cell Regul. 1990, 31, 181–221. [Google Scholar] [PubMed]
- Fukunaga, K.; Miyamoto, E. A working model of CaM kinase II activity in hippocampal long-term potentiation and memory. Neurosci. Res. 2000, 38, 3–17. [Google Scholar] [CrossRef]
- Schulman, H.; Hanson, P.I. Multifunctional Ca2+/calmodulin dependent protein kinase. Neurochem. Res. 1993, 18, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Yamamoto, H.; Fukunaga, K.; Miyakawa, T.; Miyamoto, E. Identification of the isoforms of Ca2+/calmodulin-dependent protein kinase II in rat astrocytes and their subcellular localization. J. Neurochem. 2000, 74, 2558–2567. [Google Scholar] [CrossRef]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Bayer, K.U. CaMKII regulation in information processing and storage. Trends Neurosci. 2012, 35, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W. CaMKII: Claiming center stage in postsynaptic function and organization. Neuron 2014, 81, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Stoppini, L.; Miyamoto, E.; Muller, D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1993, 268, 7863–7867. [Google Scholar] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, S.J.; Hudmon, A.; Waxham, M.N.; Stoops, J.K. Three-dimensional reconstructions of calcium/calmodulin-dependent (CaM) kinase IIalpha and truncated CaM kinase IIalpha reveal a unique organization for its structural core and functional domains. J. Biol. Chem. 2000, 275, 14354–14359. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Meyer, T.; Stryer, L.; Schulman, H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 1994, 12, 943–956. [Google Scholar] [CrossRef]
- Miller, P.; Zhabotinsky, A.M.; Lisman, J.E.; Wang, X.J. The stability of a stochastic CaMKII switch: Dependence on the number of enzyme molecules and protein turnover. PLoS Biol. 2005, 3, e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocke, L.; Srinivasan, M.; Schulman, H. Developmental and regional expression of multifunctional Ca2+/calmodulin-dependent protein kinase isoforms in rat brain. J. Neurosci. 1995, 15, 6797–6808. [Google Scholar] [PubMed]
- Tobimatsu, T.; Kameshita, I.; Fujisawa, H. Molecular cloning of the cDNA encoding the third polypeptide (gamma) of brain calmodulin-dependent protein kinase II. J. Biol. Chem. 1988, 263, 16082–16086. [Google Scholar] [PubMed]
- Mayer, P.; Möhlig, M.; Schatz, H.; Pfeiffer, A. New isoforms of multifunctional calcium/calmodulin-dependent protein kinase II. FEBS Lett. 1993, 333, 315–318. [Google Scholar] [CrossRef]
- Srinivasan, M.; Edman, C.F.; Schulman, H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J. Cell Biol. 1994, 126, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Edman, C.F.; Schulman, H. Identification and characterization of B-CaM kinase and C-CaM kinase from rat heart, two new multifunctional Ca2+/calmodulin-dependent protein kinase isoforms. Biochim. Biophys. Acta 1994, 1221, 89–101. [Google Scholar] [CrossRef]
- Heist, E.K.; Srinivasan, M.; Schulman, H. Phosphorylation at the nuclear localization signal of Ca2+/calmodulin-dependent protein kinase II blocks its nuclear targeting. J. Biol. Chem. 1998, 273, 19763–19771. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.T. Protein phosphatase 1: Targeted in many directions. J. Cell Sci. 2002, 115, 241–256. [Google Scholar] [PubMed]
- Cohen, P.T. Two isoforms of protein phosphatase 1 may be produced from the same gene. FEBS Lett. 1988, 232, 17–23. [Google Scholar] [CrossRef]
- Dombrádi, V.; Axton, J.M.; Brewis, N.D.; da Cruz e Silva, E.F.; Alphey, L.; Cohen, P.T. Drosophila contains three genes that encode distinct isoforms of protein phosphatase 1. Eur. J. Biochem. 1990, 194, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Shima, H.; Kitagawa, Y.; Irino, S.; Sugimura, T.; Nagao, M. Identification of members of the protein phosphatase 1 gene family in the rat and enhanced expression of protein phosphatase 1alpha gene in rat hepatocellular carcinomas. Jpn. J. Cancer Res. 1990, 81, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.M.; Craig, S.P.; Spurr, N.K.; Cohen, P.T. Sequence of human protein serine/threonine phosphatase 1and localization of the gene (PPP1CC) encoding it to chromosome bands 12q24.1-q24.2. Biochim. Biophys. Acta 1993, 1178, 228–233. [Google Scholar] [CrossRef]
- Barker, H.M.; Brewis, N.D.; Street, A.J.; Spurr, N.K.; Cohen, P.T. Three genes for protein phosphatase 1 map to different human chromosomes: Sequence, expression and gene localisation of protein serine/threonine phosphatase 1beta (PPP1CB). Biochim. Biophys. Acta 1994, 1220, 212–218. [Google Scholar] [CrossRef]
- Bordelon, J.R.; Smith, Y.; Nairn, A.C.; Colbran, R.J.; Greengard, P.; Muly, E.C. Differential localization of protein phosphatase-1alpha, beta, and gamma1 isoforms in primate prefrontal cortex. Cereb. Cortex. 2005, 15, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.E.; Zhabotinsky, A.M. Model of synaptic memory: A CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 2001, 31, 191–201. [Google Scholar] [CrossRef]
- Shioda, N.; Sawai, M.; Ishizuka, Y.; Shirao, T.; Fukunaga, K. Nuclear Translocation of Calcium/Calmodulin-dependent Protein Kinase IIδ3 Promoted by Protein Phosphatase-1 Enhances Brain-derived Neurotrophic Factor Expression in Dopaminergic Neurons. J. Biol. Chem. 2015, 290, 21663–21675. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Hoogenraad, C.C. The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu. Rev. Biochem. 2007, 76, 823–847. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Hoogenraad, C.C.; Rush, J.; Ramm, E.; Schlager, M.A.; Duong, D.M.; Xu, P.; Wijayawardana, S.R.; Hanfelt, J.; Nakagawa, T.; et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell Proteom. 2006, 5, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Barria, A.; Muller, D.; Derkach, V.; Griffith, L.C.; Soderling, T.R. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 1997, 276, 2042–2045. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, C.C.; da Cruz e Silva, E.F.; Greengard, P. The alpha and gamma 1 isoforms of protein phosphatase 1 are highly and specifically concentrated in dendritic spines. Proc. Natl. Acad. Sci. USA 1995, 92, 3396–3400. [Google Scholar] [CrossRef] [PubMed]
- Strack, S.; Kini, S.; Ebner, F.F.; Wadzinski, B.E.; Colbran, R.J. Differential cellular and subcellular localization of protein phosphatase 1 isoforms in brain. J. Comp. Neurol. 1999, 413, 373–384. [Google Scholar] [CrossRef]
- Blitzer, R.D.; Connor, J.H.; Brown, G.P.; Wong, T.; Shenolikar, S.; Iyengar, R.; Landau, E.M. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 1998, 280, 1940–1942. [Google Scholar] [CrossRef] [PubMed]
- Mullasseril, P.; Dosemeci, A.; Lisman, J.E.; Griffith, L.C. A structural mechanism for maintaining the ‘on-state’ of the CaMKII memory switch in the post-synaptic density. J. Neurochem. 2007, 103, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Colbran, R.J. Targeting of calcium/calmodulin-dependent protein kinase II. Biochem. J. 2004, 378, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Peti, W.; Nairn, A.C.; Page, R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 2013, 280, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.; Okamoto, K.; Nakagawa, T.; Neve, R.L.; Nagai, T.; Miyawaki, A.; Miyawaki, A.; Hashikawa, T.; Kobayashi, S.; Hayashi, Y. Visualization of synaptic Ca2+/calmodulin-dependent protein kinase II activity in living neurons. J. Neurosci. 2005, 25, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Escobedo-Lozoya, Y.; Szatmari, E.M.; Yasuda, R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 2009, 458, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Otmakhov, N.; Regmi, S.; Lisman, J.E. Fast Decay of CaMKII FRET Sensor Signal in Spines after LTP Induction Is Not Due to Its Dephosphorylation. PLoS ONE 2015, 10, e0130457. [Google Scholar] [CrossRef] [PubMed]
- Picconi, B.; Gardoni, F.; Centonze, D.; Mauceri, D.; Cenci, M.A.; Bernardi, G.; Calabresi, P.; Di Luca, M. Abnormal Ca2+-calmodulin-dependent protein kinase II function mediates synaptic and motor deficits in experimental parkinsonism. J. Neurosci. 2004, 24, 5283–5291. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Deutch, A.Y.; Colbran, R.J. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur. J. Neurosci. 2005, 22, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Baucum, A.J.; Bass, M.A.; Colbran, R.J. Association of protein phosphatase 1 gamma 1 with spinophilin suppresses phosphatase activity in a Parkinson disease model. J. Biol. Chem. 2008, 283, 14286–14294. [Google Scholar] [CrossRef] [PubMed]
- Shioda, N.; Beppu, H.; Fukuda, T.; Li, E.; Kitajima, I.; Fukunaga, K. Aberrant calcium/calmodulin-dependent protein kinase II (CaMKII) activity is associated with abnormal dendritic spine morphology in the ATRX mutant mouse brain. J. Neurosci. 2011, 31, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Jiang, Y.H.; Elgersma, Y.; Varga, A.W.; Carrasquillo, Y.; Brown, S.E.; Christian, J.M.; Mirnikjoo, B.; Silva, A.; Beaudet, A.L.; Sweatt, J.D. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci. 2003, 23, 2634–2644. [Google Scholar] [PubMed]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Snider, W.D. Functions of the neurotrophins during nervous system development: What the knockouts are teaching us. Cell 1994, 77, 627–638. [Google Scholar] [CrossRef]
- Lo, D.C. Neurotrophic factors and synaptic plasticity. Neuron 1995, 15, 979–981. [Google Scholar] [CrossRef]
- Matthews, R.P.; Guthrie, C.R.; Wailes, L.M.; Zhao, X.; Means, A.R.; McKnight, G.S. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol. Cell. Biol. 1994, 14, 6107–6116. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Enslen, H.; Myung, P.S.; Maurer, R.A. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994, 8, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.N.; Ho, H.Y.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 2006, 52, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, A.; Ogawa, Y.; Kitani, T.; Fujisawa, H.; Hagiwara, M. Calmodulin-dependent protein kinase II potentiates transcriptional activation through activating transcription factor 1 but not cAMP response element-binding protein. J. Biol. Chem. 1996, 271, 17957–17960. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Lou, L.; Maurer, R.A. Regulation of activating transcription factor-1 and the cAMP response element-binding protein by Ca2+/calmodulin-dependent protein kinases type I, II, and IV. J. Biol. Chem. 1996, 271, 3066–3073. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Fukunaga, K.; Takiguchi, M.; Ushio, Y.; Mori, M.; Miyamoto, E. Regulation of CCAAT/enhancer-binding protein family members by stimulation of glutamate receptors in cultured rat cortical astrocytes. J. Biol. Chem. 1996, 271, 23520–23527. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.; Cao, Z.; Rosenfeld, M.G. Calcium-regulated phosphorylation within the leucine zipper of C/EBP. Science 1992, 256, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.P.; Bonni, A.; Miranti, C.K.; Rivera, V.M.; Sheng, M.; Greenberg, M.E. L-type voltage-sensitive calcium channel activation stimulates gene expression by a serum response factor-dependent pathway. J. Biol. Chem. 1994, 269, 25483–25493. [Google Scholar] [PubMed]
- Kornhauser, J.M.; Cowan, C.W.; Shaywitz, A.J.; Dolmetsch, R.E.; Griffith, E.C.; Hu, L.S.; Haddad, C.; Xia, Z.; Greenberg, M.E. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 2002, 34, 221–233. [Google Scholar] [CrossRef]
- Gau, D.; Lemberger, T.; von Gall, C.; Kretz, O.; Le Minh, N.; Gass, P.; Schmid, W.; Schibler, U.; Korf, H.W.; Schütz, G. Phosphorylation of CREB Ser142 regulates light-induced phase shifts of the circadian clock. Neuron 2002, 34, 245–253. [Google Scholar] [CrossRef]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG-binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Gabel, H.W.; Hemberg, M.; Hutchinson, A.N.; Sadacca, L.A.; Ebert, D.H.; Harmin, D.A.; Greenberg, R.S.; Verdine, V.K.; Zhou, Z.; et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 2011, 72, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhong, X.; Chau, K.F.; Williams, E.C.; Chang, Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP, and spatial memory. Nat. Neurosci. 2011, 14, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Li, X.; Cooper, N.G. CaMKIIαB mediates a survival response in retinal ganglion cells subjected to a glutamate stimulus. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3854–3863. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Groth, R.D.; Cohen, S.M.; Emery, J.F.; Li, B.; Hoedt, E.; Zhang, G.; Neubert, T.A.; Tsien, R.W. γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 2014, 159, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar]
- Gonzales, M.L.; LaSalle, J.M. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep. 2010, 12, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Dani, V.S.; Chang, Q.; Maffei, A.; Turrigiano, G.G.; Jaenisch, R.; Nelson, S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12560–12565. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.; Young, J.; Yuva-Paylor, L.; Spencer, C.; Antalffy, B.; Noebels, J.; Armstrong, D.; Paylor, R.; Zoghbi, H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 2002, 35, 243–254. [Google Scholar] [CrossRef]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG-binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Kamata, A.; Takeuchi, Y.; Fukunaga, K. Identification of the isoforms of Ca2+/calmodulin-dependent protein kinase II and expression of brain-derived neurotrophic factor mRNAs in the substantia nigra. J. Neurochem. 2006, 96, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Fukunaga, K.; Miyamoto, E. Activation of nuclear Ca2+/calmodulin-dependent protein kinase II and brain-derived neurotrophic factor gene expression by stimulation of dopamine D2 receptor in transfected NG108–15 cells. J. Neurochem. 2002, 82, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, R.; Hori, H.; Ikenouchi-Sugita, A.; Umene-Nakano, W.; Katsuki, A.; Hayashi, K.; Atake, K.; Tomita, M.; Nakamura, J. Aripiprazole altered plasma levels of brain-derived neurotrophic factor and catecholamine metabolites in first-episode untreated Japanese schizophrenia patients. Hum. Psychopharmacol. 2012, 27, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 1999, 270, 45–48. [Google Scholar] [CrossRef]
- Parain, K.; Murer, M.G.; Yan, Q.; Faucheux, B.; Agid, Y.; Hirsch, E.; Raisman-Vozari, R. Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport 1999, 10, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Hyman, C.; Hofer, M.; Barde, Y.A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Murer, M.G.; Yan, Q.; Raisman-Vozari, R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 2001, 63, 71–124. [Google Scholar] [CrossRef]
- Woodgett, J.R.; Davison, M.T.; Cohen, P. The calmodulin-dependent glycogen synthase kinase from rabbit skeletal muscle. Purification, subunit structure and substrate specificity. Eur. J. Biochem. 1983, 136, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Brocke, L.; Chiang, L.W.; Wagner, P.D.; Schulman, H. Functional implications of the subunit composition of neuronal CaM kinase II. J. Biol. Chem. 1999, 274, 22713–22722. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Schulman, H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu. Rev. Biochem. 1992, 61, 559–601. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Teruel, M.N.; Subramanian, K.; Meyer, T. CaMKIIβ functions as an F-actin targeting module that localizes CaMKIIα/β heterooligomers to dendritic spines. Neuron 1998, 21, 593–606. [Google Scholar] [CrossRef]
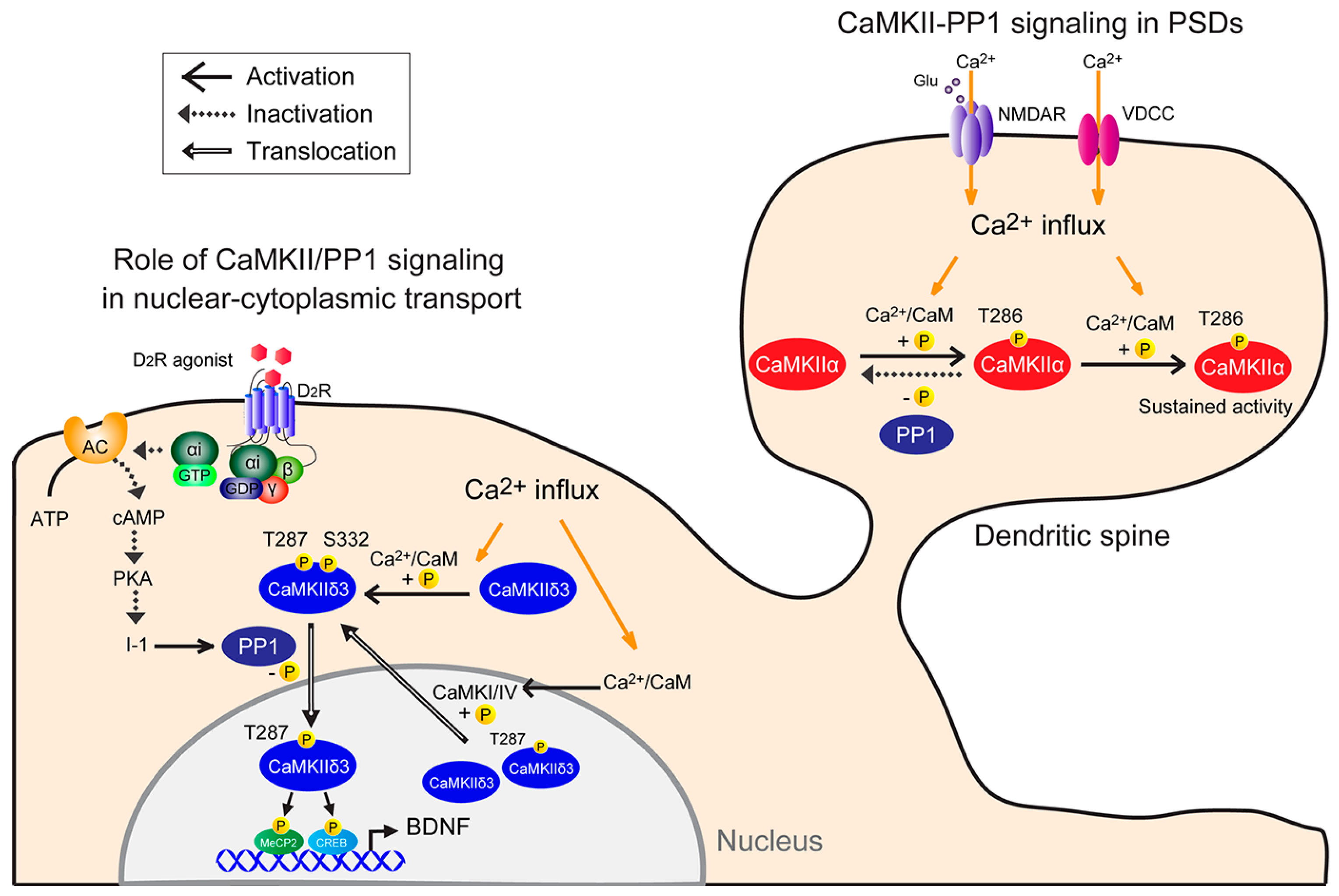
Figure 1.
Model of neuronal CaMKII-PP1 signaling. (1) CaMKII-PP1 signaling in PSDs: CaMKII is simultaneously bound by Ca2+/CaM following a Ca2+ stimulus. In this condition, one subunit acts as a substrate for the other, resulting in Thr286 phosphorylation. Once that subunit is phosphorylated, subsequent phosphorylation within the holoenzyme is more likely to occur, as Ca2+ levels required for the second phosphorylation are lower than those required for the initial phosphorylation (sustained activity). Thus, CaMKII remains active, even when basal Ca2+ levels are re-established, until it is dephosphorylated by PP1. CaMKII activity is sustained if the number of phosphorylated subunits exceeds a threshold and the phosphorylation rate exceeds the dephosphorylation rate. (2) Role of CaMKII/PP1 signaling in nuclear-cytoplasmic transport: Under basal conditions, CaMKIIδ3 is autonomously active in part due to spontaneous neuronal activity. Cytoplasmic CaMKIIδ3 is autophosphorylated, and D2R-mediated PP1 activation mediates CaMKIIδ3 dephosphorylation at Ser332. For example, stimulation with a dopamine D2R agonist increases PP1 activity by inactivating the cAMP/PKA/inhibitor 1 (I-1) pathway, and in turn PP1 dephosphorylates CaMKIIδ3 at Ser332 in the cytoplasm, enabling its nuclear translocation. Thereafter, nuclear CaMKII3 phosphorylates transcription factors, including MeCP2 and CREB, increasing BDNF expression. Depolarization causes Ca2+ entry into neurons through NMDA receptors or voltage-dependent calcium channels and promotes CaMKIIδ3 autophosphorylation at Thr287 and Ser332 in the cytosol. Conversely, nuclear CaMKI or CaMKIV activity may promote CaMKIIδ3 nuclear export via Ser332 phosphorylation.
Figure 1.
Model of neuronal CaMKII-PP1 signaling. (1) CaMKII-PP1 signaling in PSDs: CaMKII is simultaneously bound by Ca2+/CaM following a Ca2+ stimulus. In this condition, one subunit acts as a substrate for the other, resulting in Thr286 phosphorylation. Once that subunit is phosphorylated, subsequent phosphorylation within the holoenzyme is more likely to occur, as Ca2+ levels required for the second phosphorylation are lower than those required for the initial phosphorylation (sustained activity). Thus, CaMKII remains active, even when basal Ca2+ levels are re-established, until it is dephosphorylated by PP1. CaMKII activity is sustained if the number of phosphorylated subunits exceeds a threshold and the phosphorylation rate exceeds the dephosphorylation rate. (2) Role of CaMKII/PP1 signaling in nuclear-cytoplasmic transport: Under basal conditions, CaMKIIδ3 is autonomously active in part due to spontaneous neuronal activity. Cytoplasmic CaMKIIδ3 is autophosphorylated, and D2R-mediated PP1 activation mediates CaMKIIδ3 dephosphorylation at Ser332. For example, stimulation with a dopamine D2R agonist increases PP1 activity by inactivating the cAMP/PKA/inhibitor 1 (I-1) pathway, and in turn PP1 dephosphorylates CaMKIIδ3 at Ser332 in the cytoplasm, enabling its nuclear translocation. Thereafter, nuclear CaMKII3 phosphorylates transcription factors, including MeCP2 and CREB, increasing BDNF expression. Depolarization causes Ca2+ entry into neurons through NMDA receptors or voltage-dependent calcium channels and promotes CaMKIIδ3 autophosphorylation at Thr287 and Ser332 in the cytosol. Conversely, nuclear CaMKI or CaMKIV activity may promote CaMKIIδ3 nuclear export via Ser332 phosphorylation.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shioda, N.; Fukunaga, K. Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. Int. J. Mol. Sci. 2018, 19, 20. https://doi.org/10.3390/ijms19010020
AMA Style
Shioda N, Fukunaga K. Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. International Journal of Molecular Sciences. 2018; 19(1):20. https://doi.org/10.3390/ijms19010020
Chicago/Turabian StyleShioda, Norifumi, and Kohji Fukunaga. 2018. "Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain" International Journal of Molecular Sciences 19, no. 1: 20. https://doi.org/10.3390/ijms19010020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.