
TLR-2 Signaling Promotes IL-17A Production in CD4+CD25+Foxp3+ Regulatory Cells during Oropharyngeal Candidiasis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
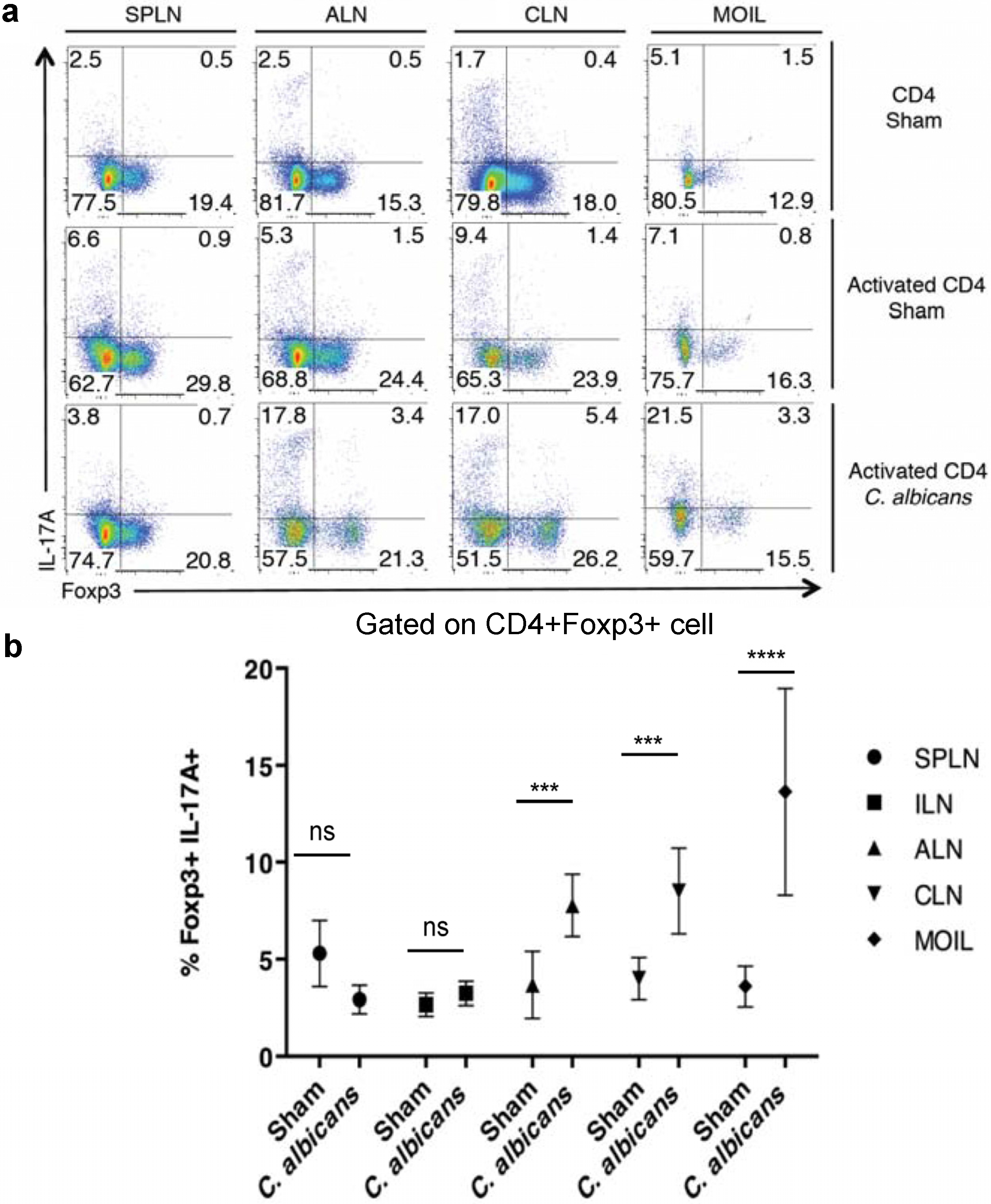
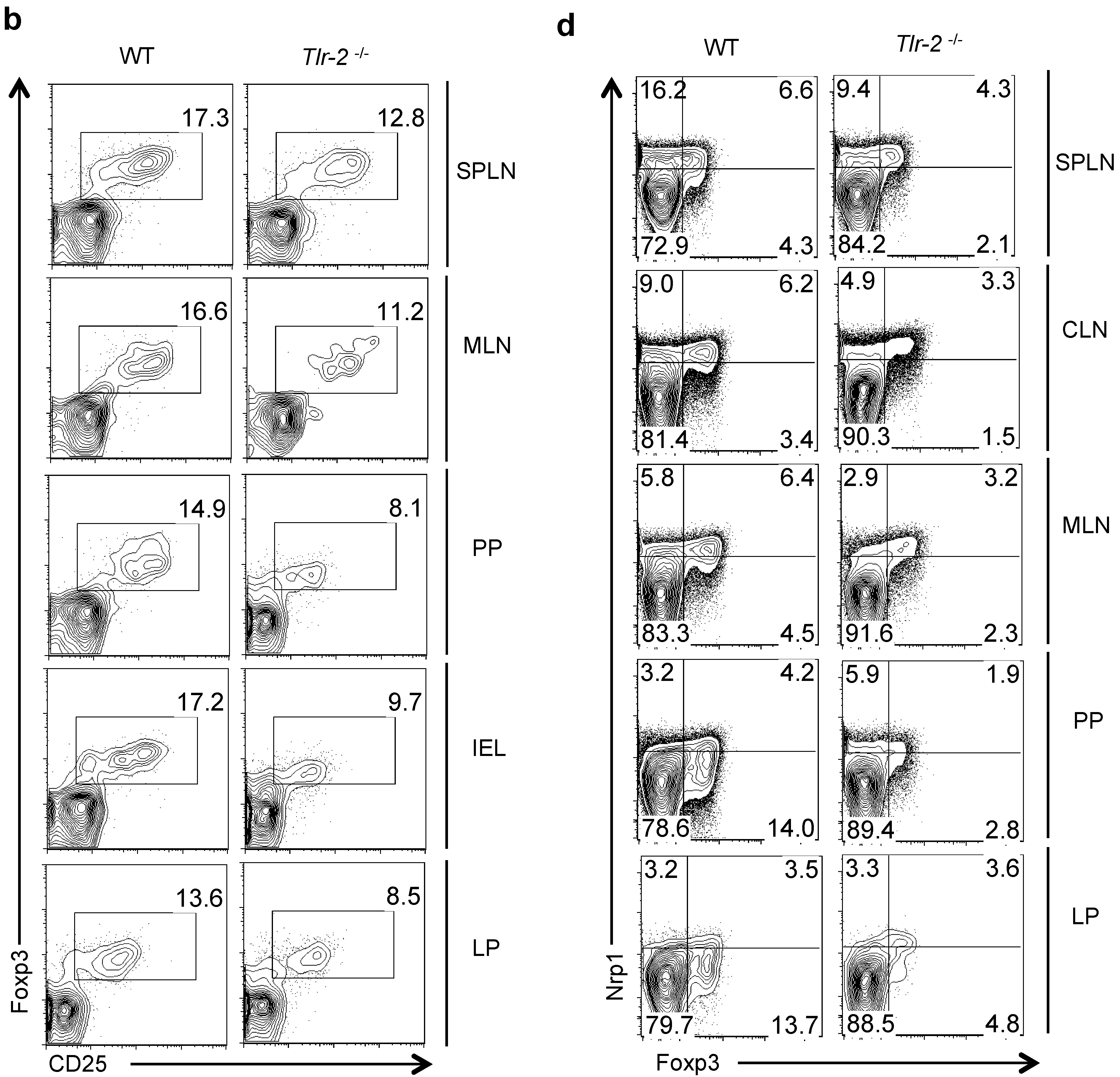
2.1. CD4+CD25+Foxp3+ Tregs Express IL-17A During Oral C. Albicans Infection and Inflammatory Bowel Disease (IBD) in Vivo

2.2. TLR-2 Ligands Directly Induce Proliferation of Tregs Independently of TCR Activation

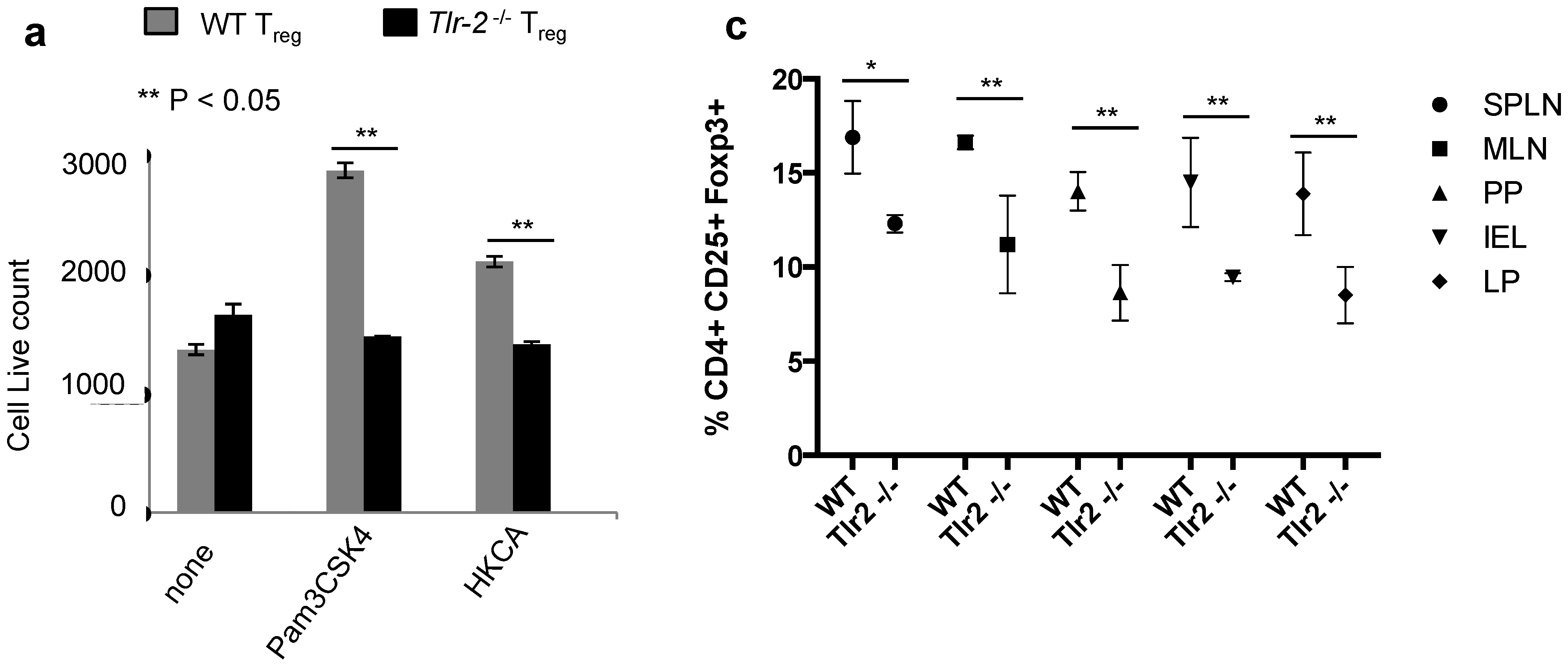
2.3. TLR-2 Ligand Mediated Proliferation in Tregs is Directly Dependent on TLR-2 Expression on Tregs


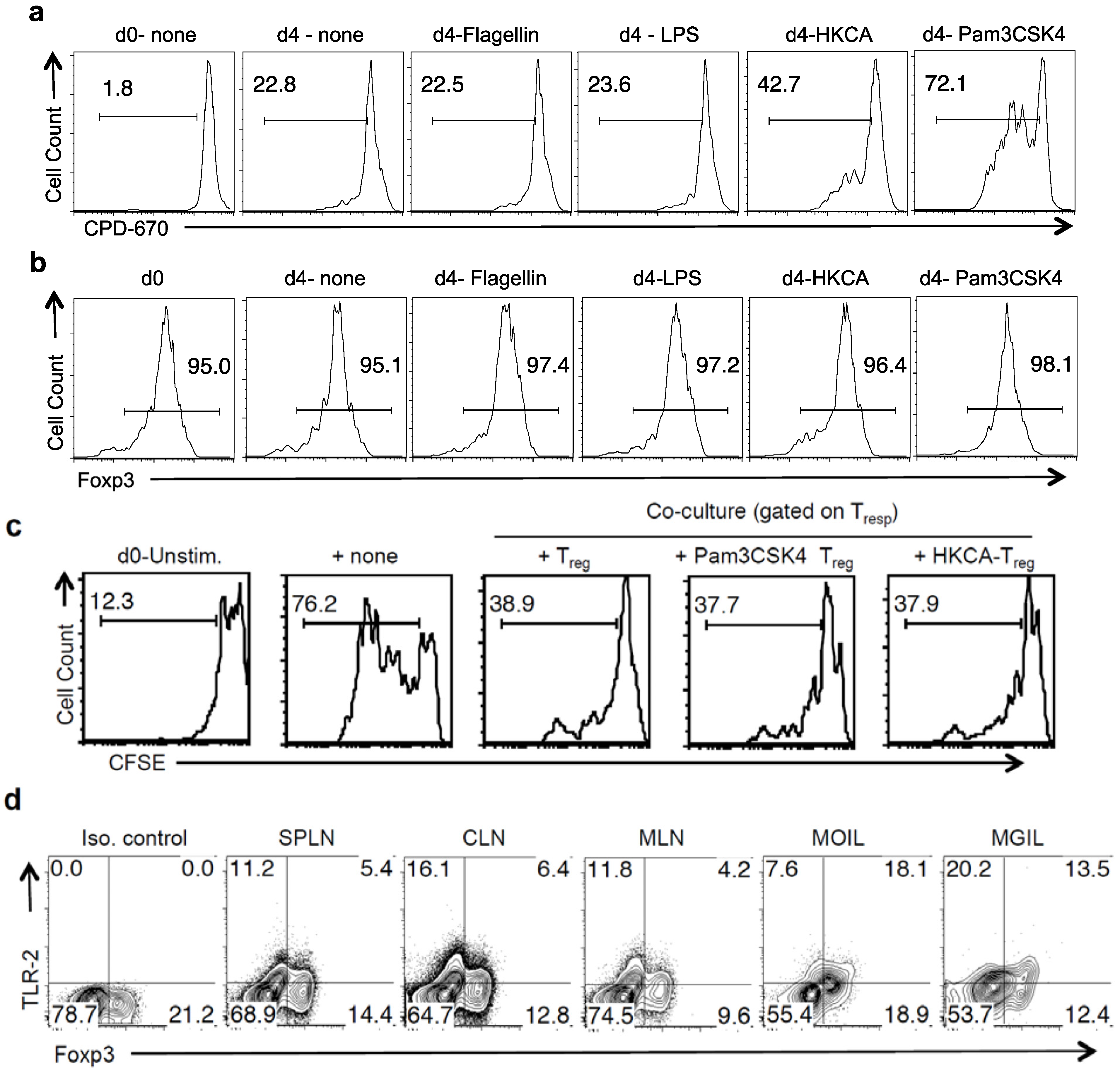
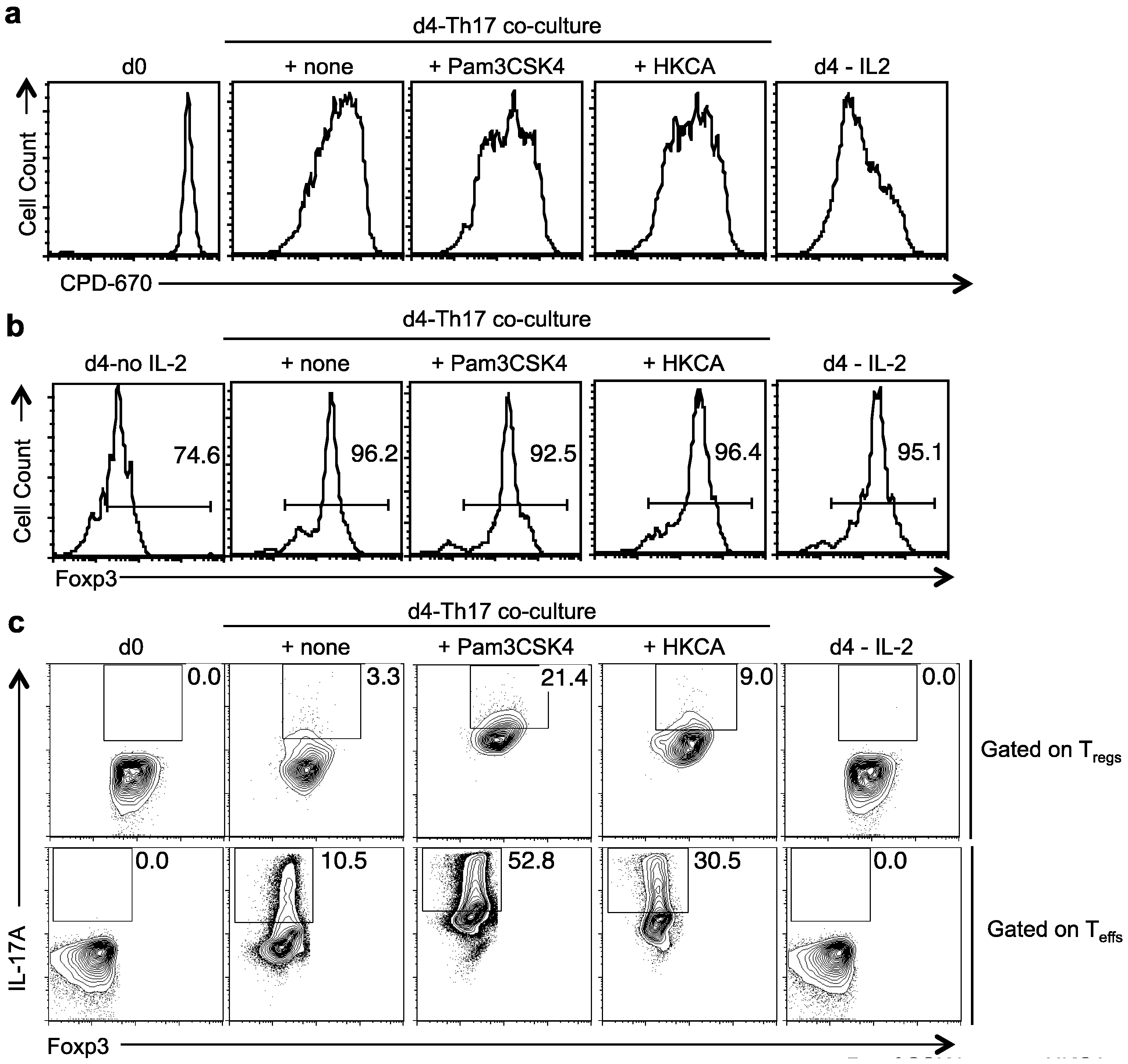
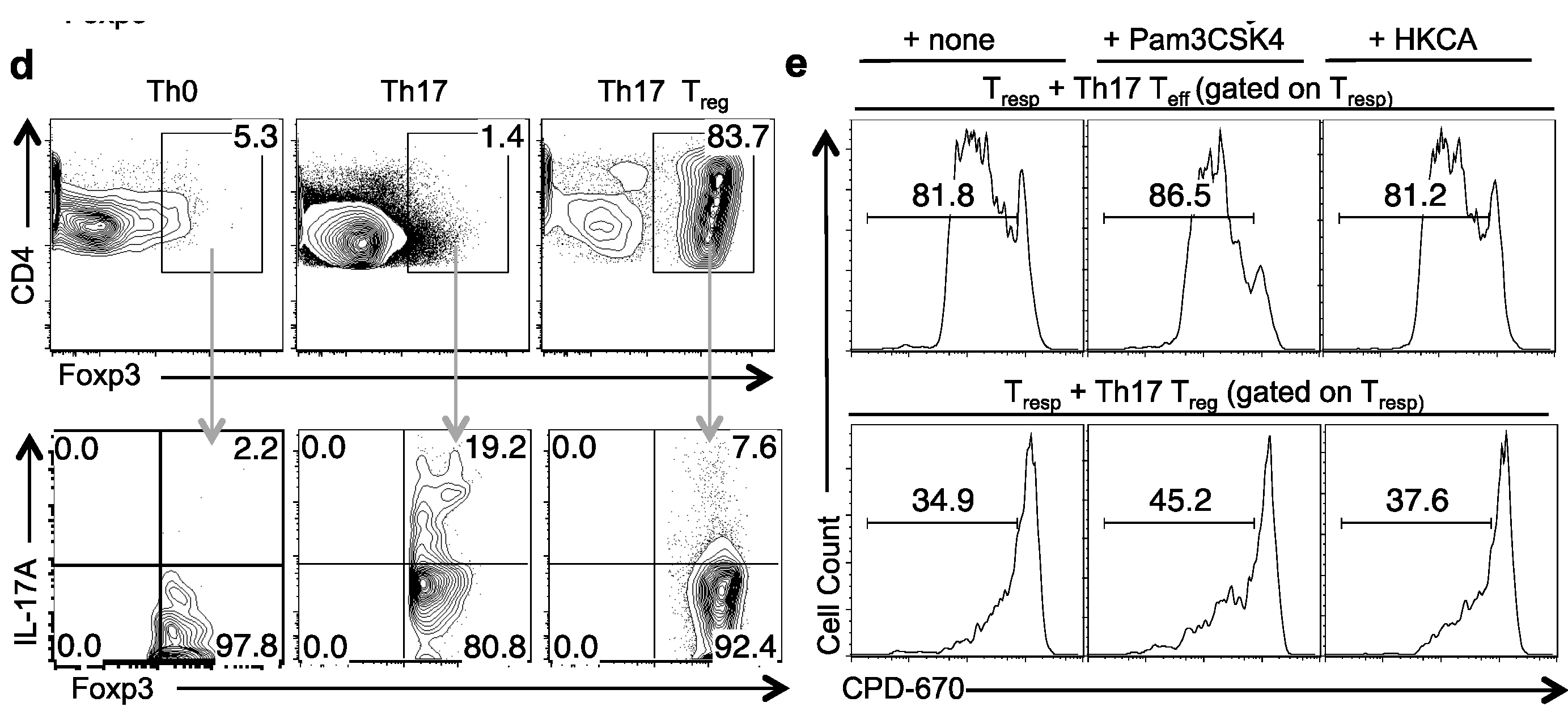
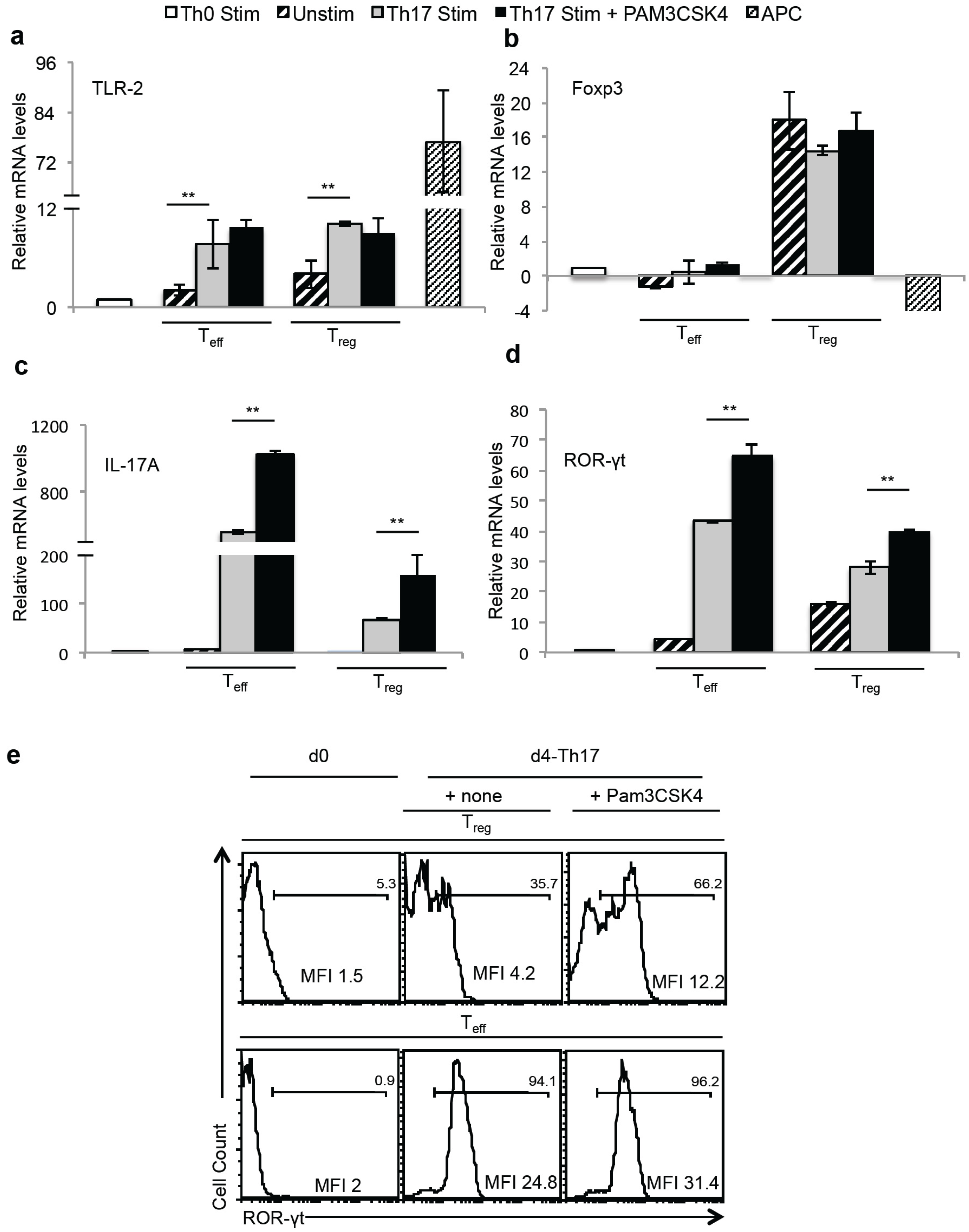
2.4. TLR-2 Ligands Did Not Reduce Foxp3 Expression, but Induced IL-17A in Foxp3+ Tregs under Th17 Conditions in Vitro



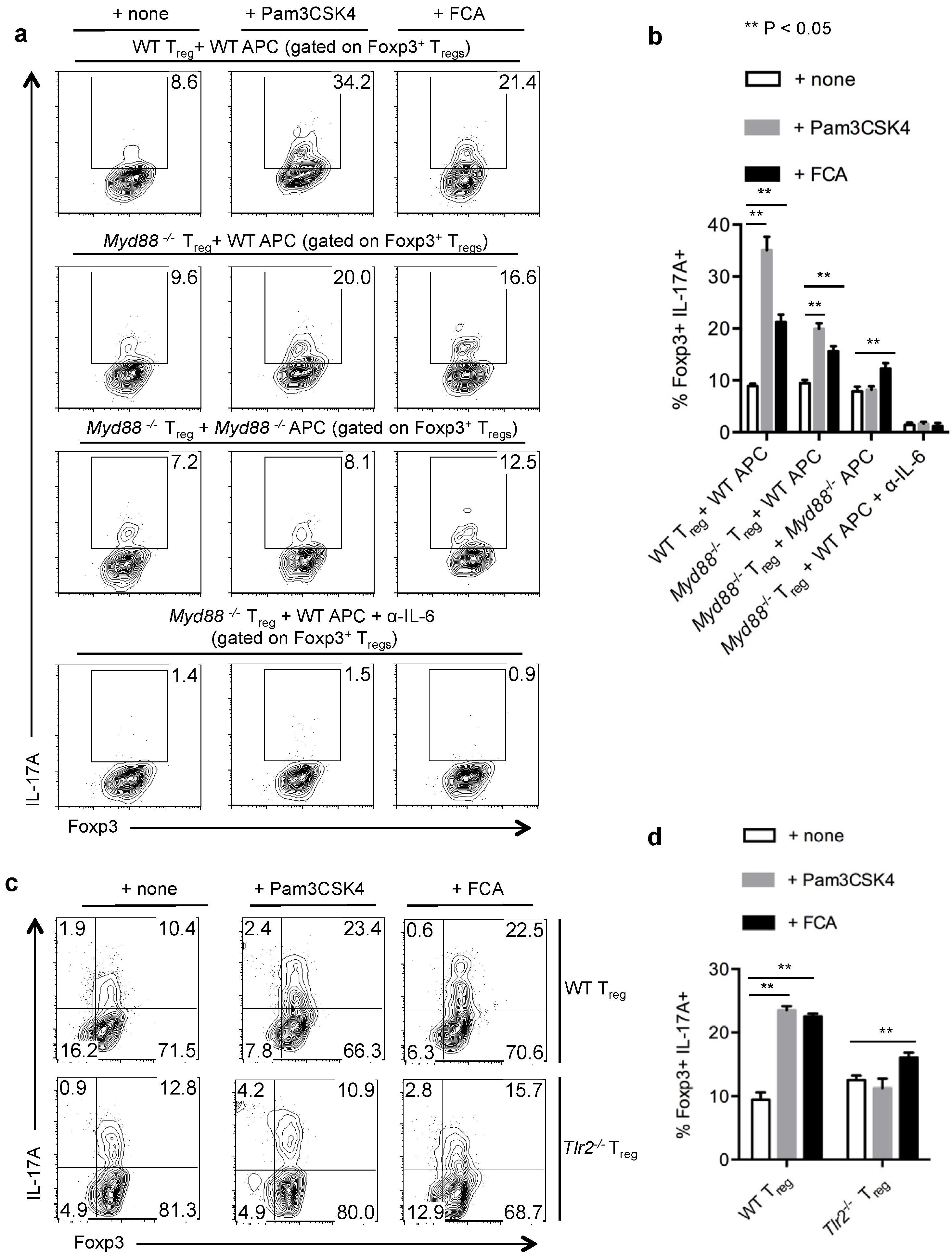
2.5. TLR-2 Ligands Induced IL-17A Production Is Only Partially Dependent on TLR-2 Expression in Tregs
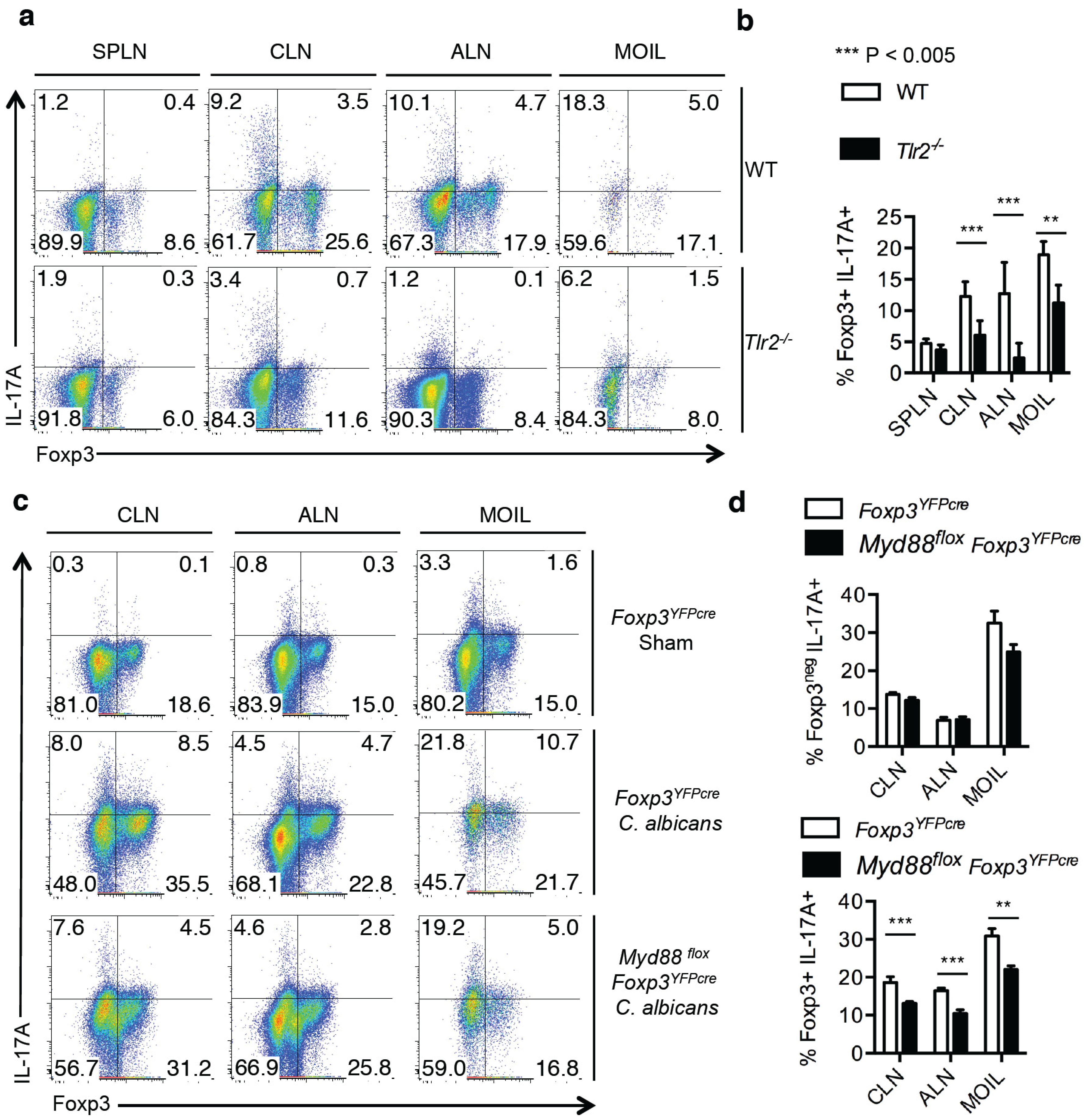
2.6. Direct TLR-2 Signaling in Tregs is Required for Maximal Induction of Foxp3+ IL-17A+ Cells during OPC in Vivo


3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Reagents and Antibodies
4.3. Cell Purification
4.4. Th17 Differentiation
4.5. Quantitative PCR (q-PCR) Analyses
4.6. C. albicans Infection in Mice
4.7. IBD Induction by Th17 Cell Transfer in Vivo
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Pandiyan, P.; Zheng, L.; Lenardo, M.J. The molecular mechanisms of regulatory T cell immunosuppression. Front. Immunol. 2011, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Rudensky, A.Y.; Campbell, D.J. In vivo sites and cellular mechanisms of t reg cell-mediated suppression. J. Exp. Med. 2006, 203, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Beriou, G.; Costantino, C.M.; Ashley, C.W.; Yang, L.; Kuchroo, V.K.; Baecher-Allan, C.; Hafler, D.A. Il-17-producing human peripheral regulatory T cells retain suppressive function. Blood 2009, 113, 4240–4249. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Zhao, H.; Yan, F.; Li, H. Il-17+foxp3+ T cells: An intermediate differentiation stage between th17 cells and regulatory T cells. J. Leukoc. Biol. 2014, 96, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, Z.; Treatman, J.; Littman, D.R.; Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 2011, 140, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Voo, K.S.; Wang, Y.H.; Santori, F.R.; Boggiano, C.; Wang, Y.H.; Arima, K.; Bover, L.; Hanabuchi, S.; Khalili, J.; Marinova, E.; et al. Identification of il-17-producing foxp3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 4793–4798. [Google Scholar] [CrossRef] [PubMed]
- Whibley, N.; Maccallum, D.M.; Vickers, M.A.; Zafreen, S.; Waldmann, H.; Hori, S.; Gaffen, S.L.; Gow, N.A.; Barker, R.N.; Hall, A.M. Expansion of foxp3(+) t-cell populations by candida albicans enhances both th17-cell responses and fungal dissemination after intravenous challenge. Eur. J. Immunol. 2014, 44, 1069–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, L.; Florek, M.; Leveson-Gower, D.B.; Sega, E.I.; Baker, J.; Smith, A.T.; Negrin, R.S. Il-17 gene ablation does not impact treg-mediated suppression of graft-versus-host disease after bone marrow transplantation. Biol. Blood Marrow Transplant. 2013, 19, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Sato, S.; Horiuchi, T.; Hoshino, K.; Takeda, K.; Dong, Z.; Modlin, R.L.; Akira, S. Cutting edge: Role of toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 2002, 169, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.J.; Koyama, S.; Nakagawa, A.; Coban, C.; Akira, S. Host innate immune receptors and beyond: Making sense of microbial infections. Cell Host Microbe 2008, 3, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors (tlrs) and their ligands. Handb. Exp. Pharmacol. 2008, 183, 1–20. [Google Scholar] [PubMed]
- Sutmuller, R.P.; Morgan, M.E.; Netea, M.G.; Grauer, O.; Adema, G.J. Toll-like receptors on regulatory T cells: Expanding immune regulation. Trends Immunol. 2006, 27, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Radler, D.; Illi, S.; Klucker, E.; Turan, E.; von Mutius, E.; Kabesch, M.; Schaub, B. Tlr2 polymorphisms influence neonatal regulatory T cells depending on maternal atopy. Allergy 2011, 66, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.M.; Pappu, B.P.; Peng, J.; Martinez, G.J.; Zhang, Y.; Chung, Y.; Ma, L.; Yang, X.O.; Nurieva, R.I.; Tian, Q.; et al. Toll-like receptor 2 signaling in cd4(+) t lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 2010, 32, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Komai-Koma, M.; Xu, D.; Liew, F.Y. Toll-like receptor 2 signaling modulates the functions of cd4+ cd25+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 7048–7053. [Google Scholar] [CrossRef] [PubMed]
- Van Maren, W.W.; Nierkens, S.; Toonen, L.W.; Bolscher, J.M.; Sutmuller, R.P.; Adema, G.J. Multifaceted effects of synthetic tlr2 ligand and legionella pneumophilia on treg-mediated suppression of T cell activation. BMC Immunol. 2011, 12, 23. [Google Scholar]
- Chen, Q.; Davidson, T.S.; Huter, E.N.; Shevach, E.M. Engagement of tlr2 does not reverse the suppressor function of mouse regulatory T cells, but promotes their survival. J. Immunol. 2009, 183, 4458–4466. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011, 332, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Pasare, C.; Medzhitov, R. Toll pathway-dependent blockade of cd4+cd25+ T cell-mediated suppression by dendritic cells. Science 2003, 299, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Hatton, R.D.; Oliver, J.; Liu, X.; Elson, C.O.; Weaver, C.T. Regulatory T cell suppression and anergy are differentially regulated by proinflammatory cytokines produced by tlr-activated dendritic cells. J. Immunol. 2004, 173, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Sutmuller, R.; Hermann, C.; van der Graaf, C.A.; van der Meer, J.W.; van Krieken, J.H.; Hartung, T.; Adema, G.; Kullberg, B.J. Toll-like receptor 2 suppresses immunity against candida albicans through induction of il-10 and regulatory T cells. J. Immunol. 2004, 172, 3712–3718. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Conti, H.R.; Zheng, L.; Peterson, A.C.; Mathern, D.R.; Hernandez-Santos, N.; Edgerton, M.; Gaffen, S.L.; Lenardo, M.J. Cd4(+)cd25(+)foxp3(+) regulatory T cells promote th17 cells in vitro and enhance host resistance in mouse candida albicans th17 cell infection model. Immunity 2011, 34, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Whibley, N.; Gaffen, S.L. Brothers in arms: Th17 and treg responses in candida albicans immunity. PLoS Pathog. 2014, 10, e1004456. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, C.; Bacci, A.; Bozza, S.; Gaziano, R.; Mosci, P.; Sharpe, A.H.; Romani, L. B7/cd28-dependent cd4+cd25+ regulatory T cells are essential components of the memory-protective immunity to candida albicans. J. Immunol. 2002, 169, 6298–6308. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Santos, N.; Huppler, A.R.; Peterson, A.C.; Khader, S.A.; McKenna, K.C.; Gaffen, S.L. Th17 cells confer long-term adaptive immunity to oral mucosal candida albicans infections. Mucosal Immunol. 2013, 6, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Bhaskaran, N.; Zhang, Y.; Weinberg, A. Isolation of T cells from mouse oral tissues. Biol. Proced. Online 2014, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory lymphocytes and intestinal inflammation. Annu. Rev. Immunol. 2009, 27, 313–338. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.L.; Gozalbo, D. Role of toll-like receptors in systemic candida albicans infections. Front. Biosci. 2009, 14, 570–582. [Google Scholar] [CrossRef]
- Gratz, I.K.; Rosenblum, M.D.; Abbas, A.K. The life of regulatory T cells. Ann. NY Acad. Sci. 2013, 1283, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Lenardo, M.J. The control of cd4+cd25+foxp3+ regulatory T cell survival. Biol. Direct 2008, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Turka, L.A.; Walsh, P.T. Il-2 signaling and cd4+ cd25+ foxp3+ regulatory T cells. Front. Biosci. 2008, 13, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kitani, A.; Fuss, I.; Strober, W. Cutting edge: Regulatory T cells induce cd4+cd25-foxp3-t cells or are self-induced to become th17 cells in the absence of exogenous tgf-beta. J. Immunol. 2007, 178, 6725–6729. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.; Zhang, N.; van der Touw, W.; Ding, Y.; Ju, W.; Bottinger, E.P.; Reid, S.P.; Levy, D.E.; Bromberg, J.S. Epigenetic regulation of foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 2009, 182, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. Cd4(+)cd25(+)foxp3(+) regulatory T cells induce cytokine deprivation-mediated apoptosis of effector cd4(+) T cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Myd88 as a bottle neck in toll/il-1 signaling. Curr. Top. Microbiol. Immunol. 2002, 270, 155–167. [Google Scholar] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Cording, S.; Wahl, B.; Kulkarni, D.; Chopra, H.; Pezoldt, J.; Buettner, M.; Dummer, A.; Hadis, U.; Heimesaat, M.; Bereswill, S.; et al. The intestinal micro-environment imprints stromal cells to promote efficient treg induction in gut-draining lymph nodes. Mucosal. Immunol. 2013, 7, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Ostman, S.; Rask, C.; Wold, A.E.; Hultkrantz, S.; Telemo, E. Impaired regulatory T cell function in germ-free mice. Eur. J. Immunol. 2006, 36, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Strauch, U.G.; Obermeier, F.; Grunwald, N.; Gurster, S.; Dunger, N.; Schultz, M.; Griese, D.P.; Mahler, M.; Scholmerich, J.; Rath, H.C. Influence of intestinal bacteria on induction of regulatory T cells: Lessons from a transfer model of colitis. Gut 2005, 54, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.P.; Chang, C.; Lai, J.J. Immune suppressive activity and lack of t helper differentiation are differentially regulated in natural regulatory T cells. J. Immunol. 2009, 183, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Song, X.; Li, B.; Greene, M.I. Foxp3 and its partners: Structural and biochemical insights into the regulation of foxp3 activity. Immunologic research 2008, 42, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; van de Veerdonk, F.L.; Lenardon, M.; Stoffels, M.; Plantinga, T.; Smeekens, S.; Rizzetto, L.; Mukaremera, L.; Preechasuth, K.; Cavalieri, D.; et al. The dectin-1/inflammasome pathway is responsible for the induction of protective t-helper 17 responses that discriminate between yeasts and hyphae of candida albicans. J. Leukoc. Biol. 2011, 90, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Rudensky, A.Y. Control of inflammation by integration of environmental cues by regulatory T cells. J. Clin. Investig. 2013, 123, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing foxp3 and bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Rudra, D.; Treuting, P.; Samstein, R.M.; Liang, Y.; Kas, A.; Rudensky, A.Y. Cd4+ regulatory T cells control th17 responses in a stat3-dependent manner. Science 2009, 326, 986–991. [Google Scholar] [CrossRef]
- Kamai, Y.; Kubota, M.; Kamai, Y.; Hosokawa, T.; Fukuoka, T.; Filler, S.G. New model of oropharyngeal candidiasis in mice. Antimicrob. Agents Chemother. 2001, 45, 3195–3197. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhaskaran, N.; Cohen, S.; Zhang, Y.; Weinberg, A.; Pandiyan, P. TLR-2 Signaling Promotes IL-17A Production in CD4+CD25+Foxp3+ Regulatory Cells during Oropharyngeal Candidiasis. Pathogens 2015, 4, 90-110. https://doi.org/10.3390/pathogens4010090
Bhaskaran N, Cohen S, Zhang Y, Weinberg A, Pandiyan P. TLR-2 Signaling Promotes IL-17A Production in CD4+CD25+Foxp3+ Regulatory Cells during Oropharyngeal Candidiasis. Pathogens. 2015; 4(1):90-110. https://doi.org/10.3390/pathogens4010090
Chicago/Turabian StyleBhaskaran, Natarajan, Samuel Cohen, Yifan Zhang, Aaron Weinberg, and Pushpa Pandiyan. 2015. "TLR-2 Signaling Promotes IL-17A Production in CD4+CD25+Foxp3+ Regulatory Cells during Oropharyngeal Candidiasis" Pathogens 4, no. 1: 90-110. https://doi.org/10.3390/pathogens4010090